the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 21 Nov 2018
| 21 Nov 2018
Large variations in iron input to an oligotrophic Baltic Sea estuary: impact on sedimentary phosphorus burial
Matthias Egger
Niels A. G. M. van Helmond
Emma Kritzberg
Daniel J. Conley
Caroline P. Slomp
Estuarine sediments are key sites for removal of phosphorus (P) from rivers and the open sea. Vivianite, an Fe(II)-P mineral, can act as a major sink for P in Fe-rich coastal sediments. In this study, we investigate the burial of P in the Öre Estuary in the northern Baltic Sea. We find much higher rates of P burial at our five study sites (up to ∼0.145 ) when compared to more southern coastal areas in the Baltic Sea with similar rates of sedimentation. Detailed study of the sediment P forms at our site with the highest rate of sedimentation reveals a major role for P associated with Fe and the presence of vivianite crystals below the sulfate methane transition zone. By applying a reactive transport model to sediment and porewater profiles for this site, we show that vivianite may account for up to ∼40 % of total P burial. With the model, we demonstrate that vivianite formation is promoted in sediments with a low bottom water salinity and high rates of sedimentation and Fe oxide input. While high rates of organic matter input are also required, there is an optimum rate above which vivianite formation declines. Distinct enrichments in sediment Fe and sulfur at depth in the sediment are attributed to short periods of enhanced input of riverine Fe and organic matter. These periods of enhanced input are linked to variations in rainfall on land and follow dry periods. Most of the P associated with the Fe in the sediment is likely imported from the adjacent eutrophic Baltic Proper. Our work demonstrates that variations in land-to-sea transfer of Fe may act as a key control on burial of P in coastal sediments. Ongoing climate change is expected to lead to a decrease in bottom water salinity and contribute to continued high inputs of Fe oxides from land, further promoting P burial as vivianite in the coastal zone of the northern Baltic Sea. This may enhance the role of this oligotrophic area as a sink for P imported from eutrophic parts of the Baltic Sea.
- Article
(5372 KB) - Full-text XML
-
Supplement
(3219 KB) - BibTeX
- EndNote





Phosphorus (P) is an important nutrient for primary producers. Burial of reactive P (i.e., bioavailable P) in coastal sediments can permanently remove P from the water column (Delaney, 1998; Froelich et al., 1982; Ruttenberg, 2003). This removal allows coastal systems to act as filters for P, reducing the flux of land-derived P to the open sea (Asmala et al., 2017; Bouwman et al., 2013; Froelich, 1988), and as sinks of P imported from the open sea (Asmala et al., 2017). The major sedimentary phases contributing to the removal of reactive P in coastal sediments are organic P, iron-bound P and authigenic calcium (Ca)-bound P (Ruttenberg and Berner, 1993; Slomp et al., 1996a).
In most coastal systems, organic matter is a major carrier of P to the sediment (Slomp, 2011). The subsequent microbial degradation of part of the organic matter can lead to elevated concentrations of phosphate () in the porewater and precipitation of authigenic P minerals (Froelich et al., 1988; Ruttenberg, 2003). While, until recently, carbonate fluorapatite was thought to act as the major sink of P in coastal systems (Ruttenberg and Berner, 1993; Slomp et al., 1996b; Ruttenberg, 2003), increasing evidence points towards a significant role for the Fe(II)-phosphate mineral vivianite (Dijkstra et al., 2016; Egger et al., 2015a; Li et al., 2015; Slomp et al., 2013).
Vivianite can form in sediments where the supply of sulfate () is low relative to the input of Fe(oxyhydr)oxides (henceforth termed Fe oxides). As a consequence, not all Fe oxides are converted to Fe sulfides (FeS and FeS2; Ruttenberg, 2003) and Fe oxides can continue to dissolve at depth through Fe reduction coupled to organic matter degradation or anaerobic oxidation of methane (Beal et al., 2009; Egger et al., 2015b; Ettwig et al., 2016). In coastal sediments, conditions for the formation of vivianite are especially favorable below the sulfate methane transition zone (SMTZ; Egger et al., 2015a; Hsu et al., 2014; März et al., 2008; Roden and Edmonds, 1997). Here, both porewater Fe2+ and can accumulate in the absence of sulfide and precipitate as vivianite (Dijkstra et al., 2016; Egger et al., 2015a; Ruttenberg, 2003). Therefore, sediments with high rates of sedimentation and inputs of organic matter and Fe oxides and with an SMTZ that is close to the sediment–water interface are thought to be particularly conducive to vivianite formation (Egger et al., 2015a; Slomp et al., 2013).
Recent studies of diagenesis in sediments of the northern Baltic Sea indicate that variations in the input of organic matter and Fe oxides may contribute to temporal variations in vivianite formation in coastal sediments (Egger et al., 2015a; Rooze et al., 2016). While Egger et al. (2015a) attributed this to eutrophication, the large magnitude of the change in input of organic matter and of Fe required to describe these data with a reactive transport model (factors of 15 and 2, respectively; Rooze et al., 2016) suggests that additional processes are active. We hypothesize that variations in river input of both organic matter and Fe, linked to variations in river discharge, play a role. This hypothesis is based on observed changes in Fe dynamics in soils and rivers in Finland (Sarkkola et al., 2013). More specifically, dry periods may lower groundwater levels exposing Fe minerals in previously reducing soil layers to oxidation (Laudon et al., 2011). After dry periods, the freshly formed Fe oxides can be reduced again, after which Fe can be mobilized as Fe2+ and transported to rivers and the coastal zone (Sarkkola et al., 2013). While some of the Fe2+ will be oxidized to form amorphous and crystalline Fe oxides (Pokrovsky and Schott, 2002; Schwertmann and Taylor, 1989) and will be transported downstream in particulate form, some of the Fe2+ can also form complexes with dissolved organic compounds and remain in solution (Kuma et al., 1996; Rue and Bruland, 1995; Wu and Luther, 1995). Both the particulate and dissolved Fe and the associated organic matter may be transported to the coastal zone through river flow where they may impact P burial.
Future climate change is expected to alter precipitation patterns in northern Europe and change both the magnitude and seasonality of river discharge (Harley et al., 2006). In the northern Baltic Sea, an increase in the input of freshwater is expected over the coming century (Meier et al., 2006). Such a change may further promote vivianite formation by reducing the availability of (Hartzell et al., 2017; Jordan et al., 2008). Environmental change may also alter riverine input of Fe. In the northern Baltic Sea, the input of dissolved Fe from rivers has increased over the past decades (Björnerås et al., 2017; Kritzberg and Ekström, 2012; Sarkkola et al., 2013). The associated increased input of Fe oxides to coastal sediments may also enhance vivianite formation.

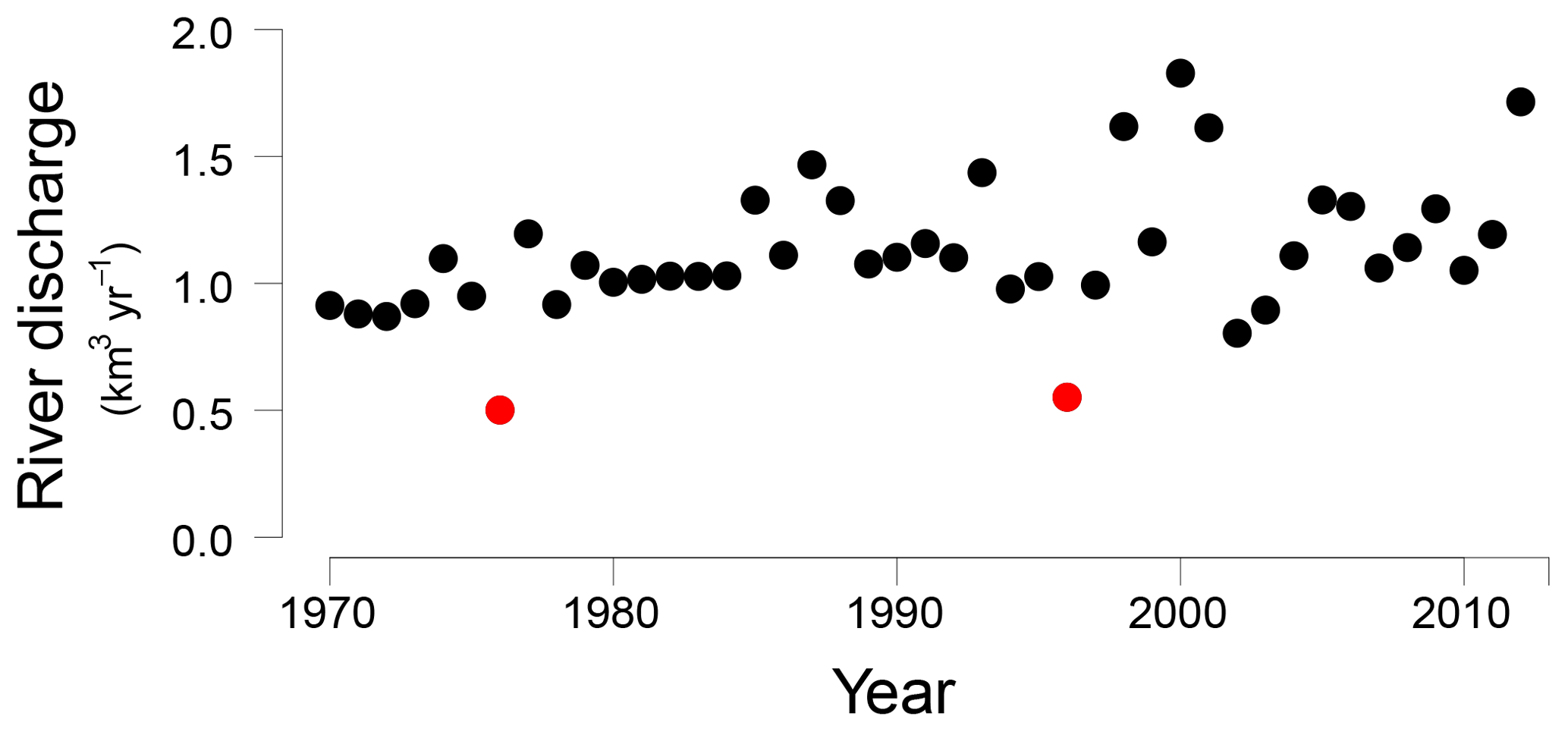
Figure 2Öre River discharge (km3 yr−1) from 1970 to 2012 (Swedish Meteorological and Hydrological Institute, http://SMHI.se, last access: 9 May 2016). The red dots indicate two years with an unusually low river runoff, 1976 and 1996, respectively.
In this study, we investigate the burial of P in the Öre Estuary in the northern Baltic Sea, with a specific focus on the factors contributing to vivianite formation. Porewater and sediment geochemical depth profiles for five sites characterized by varying rates of sedimentation are presented. We apply a reactive transport model to the data for one of the sites and investigate how vivianite formation and P burial in the sediment responds to changes in (1) bottom water salinity, (2) sedimentation rate, (3) the input of Fe oxides and (4) the input of organic matter. We also discuss the expected changes in the burial of P in coastal sediments upon ongoing climate change. Our model results suggest that vivianite will become a more important sink for P in the coastal zone of the Baltic Sea in future because of the expected decline in salinity and the continued high input of Fe from land.
2.1 Study area and sampling
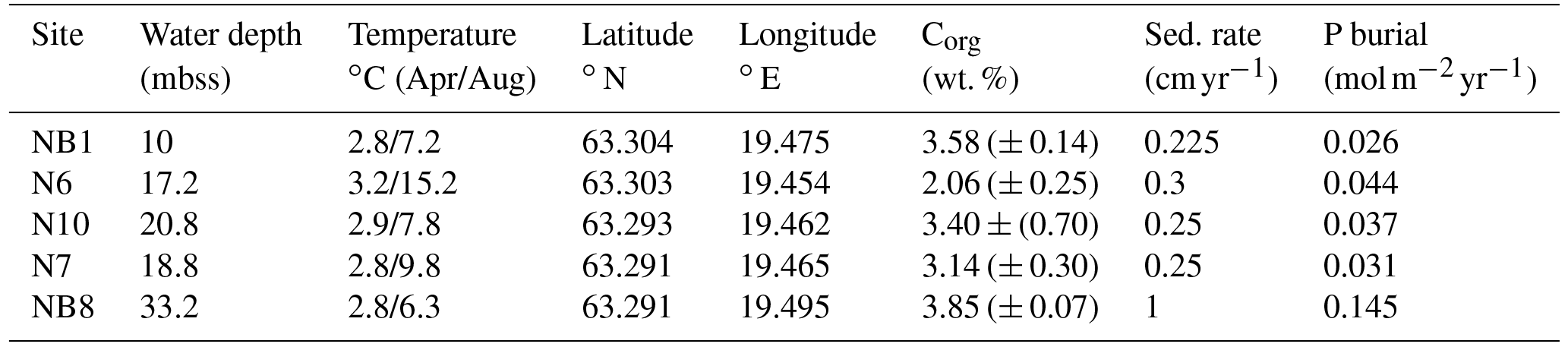
The Öre Estuary is located along the Swedish coast in the Bothnian Sea, in the northern Baltic Sea (Fig. 1). The estuary is oligotrophic and has a surface area of approximately 70 km2, a mean depth of 10 m and a bottom water salinity of ca. 5 (http://SMHI.se, last access: 8 September 2016). The estuary is fed by the Öre River, which has a strongly varying rate of annual discharge (Fig. 2). While surface water salinity varies with time and distance from the river, there is no salinity gradient in the bottom water of this estuary. The spatial extent of the particle plume from the river varies with time but is known to reach all of our sample sites (Malmgren and Brydsten, 1992). In this region, the spring flood (April, May) is the major annual hydrological event. The spring flood results in a brief period of enhanced water flow in streams and rivers (Björkvald et al., 2008; Hölemann et al., 2005) and export of Fe and terrestrial organic matter to the coastal zone (Algesten et al., 2006; Rember and Trefry, 2004). During high flow, total and dissolved Fe and Mn in streams can increase by a factor of up to 10, with concentrations being highly variable between years (Björkvald et al., 2008).
This study focuses on sediments from five sites in the Öre Estuary (Fig. 1b), with water depths varying from 10 to 33 m (Table 1). Sediments at all sites are fine-grained and rich in organic matter (Hellemann et al., 2017; Table 1). The major macrofauna species in the area are the mollusk Limecola balthica (formerly named Macoma balthica), the amphipod Monoporeia affenis, the spionid Marenzelleria spp. and the isopod Saduria entomon (Fig. S1). Marenzelleria first appeared in 1995, but only became abundant after 2002/2003, with densities at our study sites ranging from 187 to 780 ind m−2 (Kauppi et al., 2015). The density of Marenzelleria spp. in the sediment is water depth dependent with maximum densities at water depths between 30 and 50 m and lower densities both above and below (Kauppi et al., 2015).
Sediment was collected during two field campaigns with R/V Lotty in April and August 2015 using a Gemini gravity corer (8 cm inner diameter). In April, three cores were taken per site. The first core was used for porewater and solid-phase analyses, the second core was used for methane (CH4) sampling and the third core for sediment dating using 210Pb. In August, two cores were collected per site for porewater analyses and for CH4. At site NB8, a third and a fourth core were collected in August for mineral analyses and additional 210Pb measurements.
Table 1Water depth, temperature, coordinates and organic carbon at five sites sampled in the Öre Estuary in April and August 2015. Depth unit mbss is meters below sea surface. Bottom water temperatures were derived from CTD data. Total organic carbon (Corg) is given as the average for the top 2 cm (n=2), with standard deviation between parentheses. Sedimentation rates for sites NB1, N6, N10 and N7 were determined on cores collected in April 2015. The sedimentation rate at site NB8 was determined on a core collected in August 2015.
Methane was sampled directly after core recovery using a core liner with pre-drilled holes with a 2.5 cm depth spacing. Samples of 10 mL were taken with cutoff syringes from each hole and immediately transferred to a 65 mL glass bottle filled with saturated salt solution. The bottles were stoppered, capped and stored upside down until analysis.
After core retrieval, two bottom water samples were taken and the cores were subsequently sliced into intervals of 1 to 4 cm under a nitrogen atmosphere at bottom water temperature (Table 1). Each sediment sample was divided over a pre-weighed glass vial for solid-phase analysis and determination of water content, and a 50 mL centrifuge tube. The glass vials were stored in nitrogen flushed gas-tight aluminum bags at −20 ∘C until analysis. The 50 mL centrifuge tubes were centrifuged at 3500 rpm for 20 min to extract porewater.
2.2 Bottom water and porewater analyses
Bottom and porewater samples were filtered through 0.45 µm pore size filters and subsampled under a nitrogen atmosphere. Subsamples were taken for analysis of , hydrogen sulfide (where H2S represents the sum of H2S, HS− and S2−), Fe, manganese (Mn), , ammonium () and dissolved inorganic carbon (DIC). Total Fe and Mn are assumed to represent Fe2+ and Mn2+; although in the latter case some Mn3+ may also be included (Madison et al., 2011). Subsamples for were stored frozen at −20 ∘C. All other subsamples were stored at 4 ∘C until analysis. Samples for were analyzed with ion chromatography (detection limit of <75 µmol L−1; average analytical uncertainty based on duplicates and triplicates <2 %). For H2S, 0.5 mL of porewater was immediately transferred into a 4 mL glass vial containing 2 mL of 2 % zinc acetate solution to trap the H2S as ZnS. Sulfide was determined spectrophotometrically by complexing of the ZnS precipitate in an acidified solution of phenylenediamine and ferric chloride (Cline, 1969). The sulfide standard was verified by titration with thiosulfate. Subsamples taken for porewater Fe2+, Mn2+ and were acidified with 10 µL 30 % Suprapur® HCl per milliliter of sample and were analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES; Spectro Arcos).
Concentrations of were determined with the modified indophenol blue method (Solorzano, 1969). Samples for DIC analyses were collected in 5 mL vials without a headspace and poisoned with 10 µL of saturated HgCl2. Analysis for DIC was performed using an AS-C3 analyzer (Apollo SciTech), consisting of an acidification and purging unit in combination with a LI-COR 7000 CO2∕H2O gas analyzer.
Samples for CH4 were prepared for measurement by injecting a 10 mL nitrogen headspace into the bottle. Subsequently, the CH4 concentrations in the headspace were determined by injection of a subsample (50–200 µL) into a Thermo Finnigan Trace GC gas chromatograph (flame ionization detector), after which calculated CH4 concentrations were corrected for sediment porosity. The average analytical uncertainty based on duplicates and triplicates was <3 % for DIC and <5 % for CH4.
2.3 Solid-phase analyses
Sediments were freeze-dried and the porosity was determined from the weight loss upon freeze-drying. Freeze-dried sediments were ground in an agate mortar under a nitrogen atmosphere and were split into subsamples that were stored either under oxic or anoxic conditions. For site NB8, the speciation of solid-phase Fe, S and P was determined on the anoxic subsamples to avoid oxidation artifacts (Kraal and Slomp, 2014; Kraal et al., 2009). All other analyses were performed on the oxic subsamples.
Samples of ca. 300 mg of sediment was decalcified with two wash steps of 1 M HCl (Van Santvoort et al., 2002) and subsequently dried, powdered and analyzed for carbon using an elemental analyzer (Fison Instruments model NA 1500 NCS). Organic carbon (Corg) contents were determined after correction for the weight loss during decalcification. The average analytical uncertainty based on duplicates and triplicates was <5 %. A second subsample of ca. 125 mg was digested in 2.5 mL of HClO4 and HNO3 (ratio 3 : 2) and 2.5 mL 40 % HF in a Teflon vessel at 90 ∘C overnight. The acid was evaporated at 140 ∘C until a gel was formed, which was subsequently dissolved in 25 mL of 4.5 % HNO3 at 90 ∘C overnight. Total elemental concentrations in the HNO3 solution of Al, Fe, Mn, P and S were determined by ICP-OES, from which total concentrations in the sediment samples were calculated. The average analytical uncertainty based on duplicates and triplicates was <3 % for all reported elements.
Sediment samples of ca. 50 mg were subjected to the four-step Fe speciation procedure of Poulton and Canfield (2005) that targets the following Fe phases: (i) carbonate-associated Fe (including siderite and ankerite), extracted for 24 h with 1 M Na acetate (pH brought to 4.5 with acetic acid); (ii) easily reducible Fe oxides and vivianite (including ferrihydrite and lepidocrocite), extracted for 24 h with 1 M hydroxylamine-HCl in 25 % v∕v acetic acid; (iii) reducible (crystalline) Fe oxides and vivianite (including goethite, hematite and akagenéite), extracted for 2 h with Na-dithionite (pH 4.8); (iv) recalcitrant Fe oxides (mainly magnetite), extracted for 2 h with 0.2 M ammonium oxalate/0.17 M oxalic acid. Samples were measured colorimetrically using the 1,10-phenanthroline method (APHA, 2005). For simplicity, fractions (ii) and (iii) were summed and henceforth referred to as total Fe oxides. Average analytical uncertainty, based on duplicates, was <5 % for all fractions.
Sediment samples of ca. 500 mg were subjected to the three-step S speciation procedure of (Burton et al., 2006, 2008) that targets the following S phases: (i) acid-volatile sulfur (AVS, i.e., mostly FeS) by addition of 2 mL of ascorbic acid and 10 mL of 6 M HCl to the sediment and trapping of the released H2S into a tube filled with 7 mL Zn-acetate; (ii) elemental sulfur (S0) by extracting overnight with 25 mL methanol; (iii) chromium-reducible sulfur (CRS, i.e., mostly FeS2) by addition of acidic chromium chloride solution and trapping of the released H2S into a tube filled with 7 mL Zn-acetate. Samples for AVS and CRS were analyzed by iodometric titration of the alkaline Zn-acetate trap. Elemental sulfur was measured colorimetrically according to Bartlett and Skoog (1954). Average analytical uncertainty, based on duplicates, was <3 % for all fractions.
Sediment samples of ca. 100 mg were subjected to the SEDEX method described by Ruttenberg (1992) as modified by Slomp et al. (1996a), but including the exchangeable P step. Five P phases were distinguished: (i) exchangeable-P, extracted for 30 min with 1 M MgCl2 (pH 8); (ii) Fe-bound P fraction (including Fe-oxide-bound P and vivianite; Dijkstra et al., 2014; Nembrini et al., 1983) extracted for 8 h with citrate-dithionite-bicarbonate (CDB; pH 7.5) followed by extraction for 30 min with 1 M MgCl2 (pH 8); (iii) authigenic Ca-P (including carbonate fluorapatite, biogenic hydroxyapatite and carbonate-bound P), extracted for 6 h with Na acetate buffer (pH 4) followed by extraction for 30 min with 1 M MgCl2 (pH 8); (iv) detrital Ca-P, extracted for 24 h with 1 M HCl; (v) organic P, extracted for 24 h with 1 M HCl after ashing at 550 ∘C for 2 h. Steps (i)–(iv) were performed under an argon atmosphere to avoid oxidation artifacts (Kraal and Slomp, 2014; Kraal et al., 2009). CDB solutions were analyzed for P with ICP-OES. All other solutions were measured colorimetrically according to Strickland and Parsons (1972). Average analytical uncertainty, based on duplicates, was <5 % for all fractions.
2.4 Sedimentation rate and P burial
Sediment accumulation rates at all 5 sites were determined from depth profiles of 210Pb for April (NB1, N6, N10 and N7) or August (NB8). 210Pb was measured on freeze dried sediment by direct gamma counting at 46.5 keV using a high-purity germanium detector (Ortec GEM-FX8530P4-RB). Self-absorption was measured directly and the detector efficiency was determined by counting a National Institute of Standards and Technology sediment standard. Excess 210Pb was calculated as the difference between the measured total 210Pb and the estimate of the supported 210Pb activity as given by 214Pb (210Pbexc = 210Pbtotal − 214Pb). Sediment accumulation rates at each site were estimated by fitting a reactive transport model (Soetaert and Herman, 2008) to the 210Pb depth profiles assuming depth dependent changes in porosity and bioturbation (Fig. S2).
Total P burial () for all sites was calculated as follows:
where Ptotal is the averaged concentration of total P (mol g−1) over the deepest 10 cm of the sediment sampled, ϕ is the porosity in the same interval (vol vol−1), sed. rate is the sedimentation rate (cm yr−1) and ρ is the density of dry sediment, 2.65 g cm−3 (Burdige, 2006).
2.5 Scanning electron microscopy (SEM)
Sediment samples from site NB8 were analyzed by SEM to determine whether large vivianite crystals (≥38 µm size) were present. Wet sediments from five depths (sediment depth intervals: 3–4, 14–16, 34–36, 49–52 and 55–58 cm) were sieved through a 38 µm mesh size sieve under an argon atmosphere with deoxygenated ultraclean water. The sieved material was washed seven times (5 min) with deoxygenated ultraclean water in a sonic bath. After washing, the samples were dried in an argon-filled glove box at ambient temperature. A subsample of the sieved and dried material was mounted on an aluminum sample holder using double sided carbon tape and subsequently coated with 0.8 mm platinum. Samples were analyzed using SEM energy dispersive X-ray spectroscopy (EDS; JCM 6000PLUS NeoScope Benchtop SEM) with 15 kV accelerating voltage using a Si/Li detector, in scanning electron mode. Measurements with EDS were performed in the 0–20 keV energy range for elemental quantification (probe current: 1 nA; acquisition time: 50 s, live time). SEM-EDS software was used to estimate the relative abundances in mol % for the major elements (oxygen, O; sodium, Na; magnesium, Mg; Al; silica, Si; P; calcium, Ca; Mn; Fe). Samples were detected and photographed in secondary electron imaging (SEI) mode. Measurements were performed with a 1 µm beam in backscattered electron imaging (BEI).
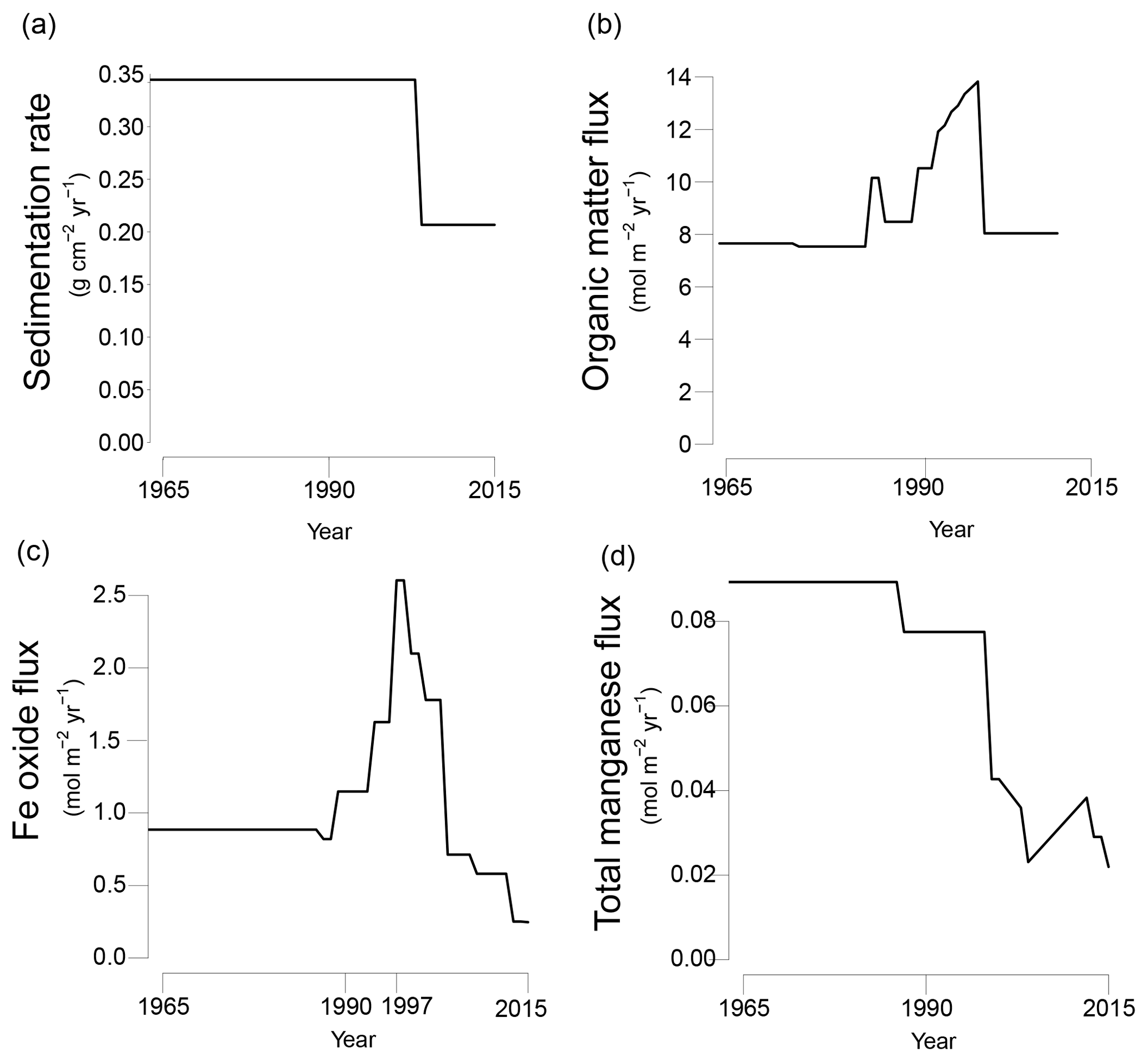
Figure 3Transient fluxes at the sediment–water interface from 1965 to 2015 as used in the reactive transport model; (a) sedimentation rate; (b) organic matter input; (c) Fe oxide input; (d) total Mn input (Mn oxide and Mn carbonate). Partitioning of different phases of organic matter, Fe oxides and manganese are shown in Fig. S4a–c.
3.1 General model description
To investigate the mechanisms that control the formation of vivianite in the Öre Estuary a reactive transport model was applied to key porewater and solid-phase depth profiles for site NB8. Site NB8 was characterized by a SMTZ close to the sediment–water interface and a relatively high sedimentation rate (Table 1). The geochemistry at site NB8 is strikingly similar to that at another site with a high sedimentation rate in the Bothnian Sea (Egger et al., 2015a, b). The model describes the mass balance of 11 dissolved and 18 particulate species (Table S1) and is a modified version of that of Rooze et al. (2016), extended here to include the sedimentary Mn cycle. This is a standard multicomponent reactive transport model based on principles outlined by, for example, Wang and Van Cappellen (1996). The large number of model components, the parameters specific for the field site, and the reaction kinetics and parameters from the literature greatly limit the degrees of freedom while fitting the model to the data (Van Cappellen and Wang, 1996). Three forms of Mn are distinguished, namely Mn oxides, Mn carbonate and Mn2+. The transformations of Mn that are included are Mn carbonate and Mn oxide formation and Mn oxide reduction coupled to either H2S, Fe2+ or CH4 oxidation (Table S2; Reactions R25–R32).
The model domain consists of a one-dimensional grid of 900 evenly distributed cells that captures the interval from the sediment–water interface to a depth of 90 cm. All chemical species are subject to biogeochemical reactions (Table S2). Solids and solutes are transported by sediment accumulation and bioturbation. Solutes are additionally transported by molecular diffusion and bioirrigation (Boudreau, 1997; Soetaert et al., 1996; Wang and Van Cappellen, 1996). Bioirrigation is modeled as a nonlocal exchange process (Boudreau, 1984; Emerson et al., 1984).
3.2 Model parameterization
The model was parameterized using data for site NB8, information from the literature, and by visually fitting modeled porewater and solid-phase depth profiles to the measured data (model constrained; Table S3). Environmental parameters such as porosity, temperature, salinity and bottom water solute concentrations were measured (Table S4; Fig. S3a). The density of dry sediment and the C : N ratio for organic matter were taken from literature (Burdige, 2006; Redfield, 1958). Bioturbation and bioirrigation coefficients were used as fitting parameters (Fig. S3b and c), taking into account when the most important bioirrigator in these sediments, Marenzelleria spp., established (Fig. S1) and the typical range in these coefficients at similar macrofaunal densities (Renz and Forster, 2013). Fluxes of solids at the sediment–water interface were model constrained. A detailed description of the reactive transport model is given in the Supplement.
3.3 Iron, manganese and phosphorus dynamics
In the model, Fe oxides and Mn oxides are assumed to consist of fractions with different crystallinities, which affect their reactivity towards organic matter and H2S (Table S1). In both cases, a highly reactive fraction (α) and a less reactive fraction (β) are assumed, whereas for Fe oxides also a refractory fraction (γ) is included. Only the α fraction of the Fe oxides and Mn oxides is susceptible to reductive dissolution linked to degradation of organic matter. This allows the β fraction to be buried below the zone of organic matter degradation (Rooze et al., 2016).
Reactive P is deposited at the sediment–water interface in the form of organic P and Fe-oxide-bound P. For organic matter three fractions are assumed: a highly reactive (α), less reactive (β) and refractory (γ) fraction, following the multi-G approach (Jørgensen, 1978; Middelburg, 1989; Westrich and Berner, 1984). The C : P ratio is 300 : 1 for all three fractions of organic matter and is model constrained. This relatively high and constant value for the C : P ratio is based on the refractory nature of the organic matter in the area (Algesten et al., 2006; Leipe et al., 2011; Stockenberg and Johnstone, 1997). The three fractions of Fe oxides are assumed to have different P : Fe ratios, with highly reactive Fe oxides having a higher P : Fe ratio then less reactive crystalline Fe oxides assuming the former can bind more P (Table S3; Gunnars et al., 2002). In the bioirrigated zone, porewater is assumed to bind to the Fe oxideβ fraction present (Fig. S3c and d) to a maximum P : Fe ratio of 0.28 (mol mol−1; Table S3). This allows for enhanced formation of Fe-oxide-bound P due to bioirrigation (Norkko et al., 2012). Nonreactive P is deposited at the sediment–water interface as detrital P, authigenic Ca–P, P bound to nonreactive Fe oxides (γ fraction) and P bound to nonreactive organic matter (γ fraction).
In the sediment, the rate of vivianite formation (Rviv) is modeled by means of Michaelis–Menten kinetics for dissolved Fe2+ and , which implies that the rate of vivianite formation depends on both porewater species and that there is a maximum rate of formation (Reed et al., 2016):
where Vmax is the maximum rate (), and are the half saturation constants, [Fe2+] and [] are the porewater concentrations of Fe2+ and .
3.4 Transient modeling scenario
The model was run to steady state for 200 years. Subsequently, temporal changes in the sedimentation rate and the input of organic matter, Fe oxides, Mn oxides and Mn carbonates were implemented to fit key porewater and solid-phase depth profiles for site NB8 (Figs. 3 and S4a–c). The modeled sedimentation rate in the first 200 years was set to 1 cm yr−1 (0.34 ), based on 210Pb dating (Fig. S2; Table 1). From 2003 onwards sedimentation was assumed to decrease to 0.6 cm yr−1 (0.21 ; Fig. 3a). The modeled organic matter deposition in the first 200 years was 7.65 . From 1987 to 2003, the deposition increased, whereas in the last 12 years the deposition decreased together with the sedimentation rate (Fig. 3b). Our modeled organic matter fluxes are at the high end of the range estimated for the region (1.1–8.2 ; Algesten et al., 2006). The qualitative trend in organic matter loading assumed in the model is in accordance with results from previous studies on eutrophication in the Bothnian Sea (Fleming-Lehtinen and Laamanen, 2012; Fleming-Lehtinen et al., 2008; Rooze et al., 2016).
The assumption is made that during strongly enhanced riverine Fe oxide input the P : Fe ratio of Fe oxides can be lower because on land the availability of is not high enough to maintain a high P : Fe ratio. This assumption is based on the dominance of natural forest and wetlands in the region (Björkvald et al., 2008). We note that the model results were very sensitive to the P : Fe ratio and that we could not model the key profiles when assuming a higher P : Fe ratio. To be able to model a transient P : Fe ratio in the Fe oxideβ fraction a fifth Fe oxide fraction, Fe oxideβ(pulse), was added to the model. This fraction has the same reactivity as Fe oxideβ towards organic matter and H2S but has a P : Fe ratio of 0. By varying the ratio between Fe oxideβ and Fe oxideβ(pulse) the P : Fe ratio of the Fe oxideβ can be adjusted over time.
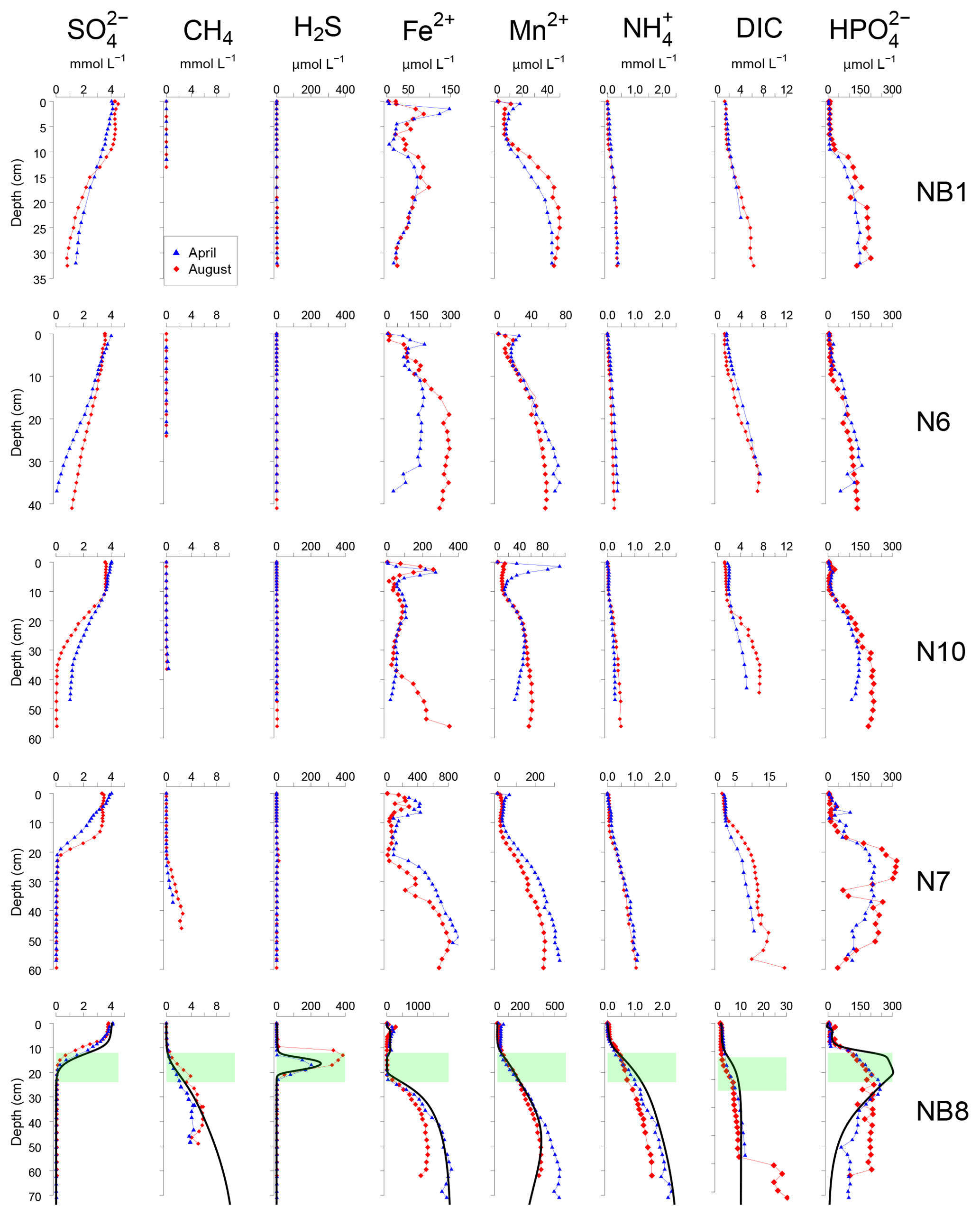
Figure 4Porewater depth profiles of , CH4, H2S, Fe2+, Mn2+, , DIC and for sites NB1, N6, N10, N7 and NB8 in April (blue) and August (red) 2015. Porewater profiles of station NB8 include model fits to the April 2015 data for the baseline scenario (black lines). The SMTZ at site NB8 is indicated by the green shaded area.
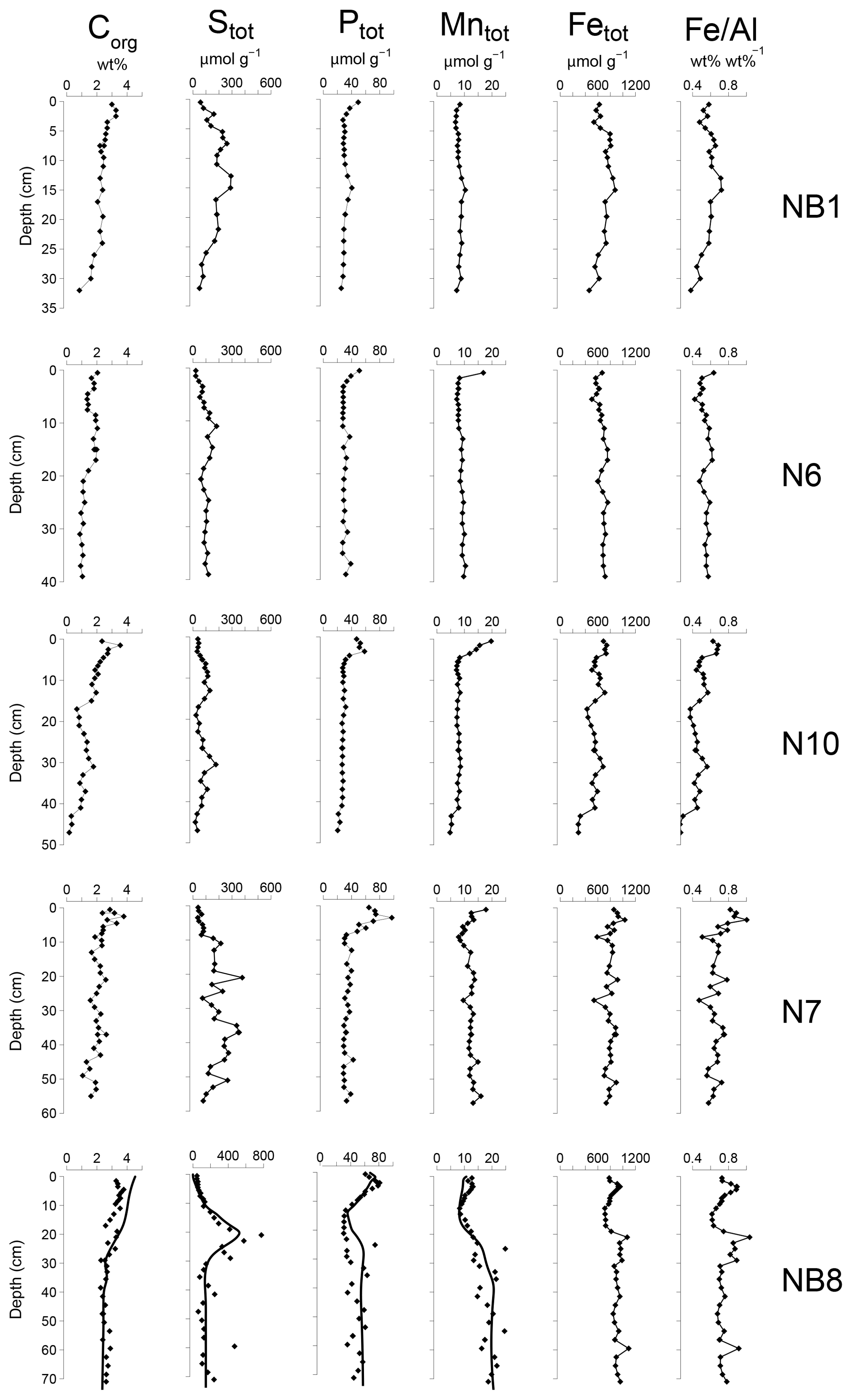
Figure 5Solid-phase depth profiles of organic carbon, total S, total P, total Mn, total Fe and Fe/Al for sites NB1, N6, N10, N7 and NB8 in April 2015. Solid-phase profiles of station NB8 include model fits to all profiles (black lines) except total Fe and Fe/Al for the baseline scenario.
In the first 200 years, the Fe oxide input was set to 0.87 . The Fe oxide input was then assumed to increase from 1988 onwards until 1997 when a maximum in incoming Fe oxides was assumed, followed by a decrease in Fe oxide input (Fig. 3c). The strong pulse-type increase in Fe oxide input at this maximum was assumed to consist of mainly the β(pulse) fraction of Fe oxides (Fig. S4b). Despite this lower P : Fe ratio, the total flux of P bound to Fe oxides was highest during this period of enhanced Fe oxide input (Fig. S4d and e). The transient scenario of Fe oxide loading compares well to qualitative trends used in a previous model study for the same region (Rooze et al., 2016). The initial Mn oxide flux was set to 0.085 (Fig. 3d). In the last 17 years of the scenario, the Mn oxide flux was set to a much lower value. The input flux of Mn carbonate was always low compared to that of Mn oxides (4–29 % of the total incoming Mn flux; Fig. S4c). Processes coupled to the presence of bioirrigating macrofauna (i.e., bioirrigation and the binding of P onto Fe oxides in the bioirrigated zone (Reaction R32 in Table S2), were only implemented in the last 12 years of the run, from 2003 onwards.
A model sensitivity analysis was performed to investigate the impact of changes in bottom water salinity, the rate of sedimentation and input of organic matter and Fe oxides on P burial rates and forms. During these sensitivity analyses the transient baseline scenario was used (Fig. 3) and one initial factor was varied per run. Salinity was varied over a range of 0 to 20. The sedimentation rate was varied from 0.25 to 2 cm yr−1. The input of organic matter was varied between −70 % and +25 %; and the input of Fe oxides was varied between −25 % and +25 % from the baseline scenario. Subsequently, a run was performed in which the input of organic matter, Fe oxides and the sedimentation rate were all changed by the same factor as the sedimentation rate to account for the role of rivers as the main source of material in the region (Algesten et al., 2006; Björkvald et al., 2008).
4.1 Porewater profiles
At all sites, depth profiles of porewater constituents showed relatively little difference between April and August 2015 (Fig. 4). Porewater decreased with depth, with a distinct SMTZ only being present in the sampled depth interval at sites N7 and NB8 (Fig. 4). Concentrations of CH4 were low at all other sites. Appreciable H2S was only present at site NB8 in the SMTZ. Dissolved Fe2+ and Mn2+ profiles generally showed a maximum near the sediment–water interface and increased concentrations below ca. 5 to 25 cm depth in the sediment. Porewater profiles of and DIC increased with depth at all sites. Porewater concentrations were low close to the sediment–water interface and increased with depth at all sites. At sites N7 and NB8, concentrations remained constant or decreased again below the SMTZ. Concentrations of Fe2+, Mn2+, and DIC were highest at depth at site NB8 when compared to the other sites.
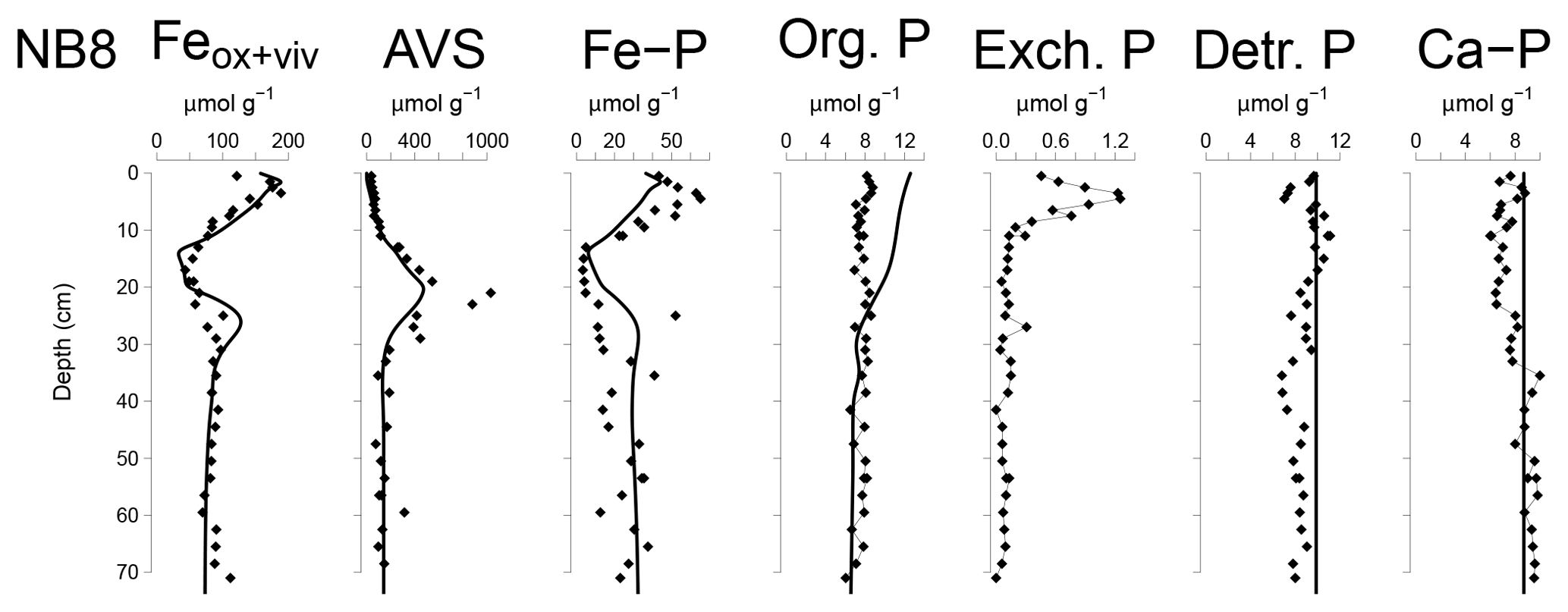
Figure 6Solid-phase depth profiles of Fe oxides, Fe-bound P (Fe–P), AVS, organic P (Org. P), exchangeable P (Exch. P), detrital Ca-P (Detr. P) and authigenic Ca-P (Ca–P) for site NB8 in April 2015. Solid-phase profiles include model fits of the baseline scenario for all profiles except exchangeable P (black lines).
4.2 Solid-phase profiles and sedimentation rate
Organic carbon contents in the surface sediment ranged between 2 and 4 wt % (Table 1) and decreased with depth at all sites (Fig. 5). Total S concentrations increased with depth in the upper 10 to 20 cm of the sediment and subsequently remained constant or varied with depth. Total P, total Mn, total Fe and Fe/Al were generally highest close to the sediment–water interface and decreased with depth at all sites except site NB8. Here, several subsurface enrichments in P and Mn were observed directly below maxima in total S, Fe and Fe/Al at depths of 21, 42 and 60 cm, respectively.
The Fe and P speciation for site NB8 reveals that the upper 10 cm of the sediment was strongly enriched in Fe oxides and Fe-bound P. Minima in both Fe oxides and Fe-bound P were found between depths of 10 and 20 cm followed by either constant (Fe oxides) or varying (Fe-bound P) concentrations at greater depth (Figs. 6 and S5). Maxima in AVS coincided with maxima in total S, total Fe and Fe/Al (Figs. 5 and 6), and minima in total Mn and Fe-bound P. Concentrations of exchangeable P were highest close to the sediment–water interface but account for only ∼2 % of total P. Concentrations of detrital P and authigenic Ca-P showed little change with depth.
Sedimentation rates at our study sites varied between 0.225 and 1 cm yr−1 (Table 1). Rates of P burial ranged from 0.026 to 0.145 (Table 1). The rates of sedimentation and P burial were highest at site NB8.
4.3 SEM-EDS
Examination of sieved sediment fractions for site NB8 with a light microscope revealed the presence of transparent to light blue crystals in sediment intervals below the SMTZ (55–58 cm). Scanning electron micrographs of the sieved fractions revealed crystals with a similar shape to that of vivianite crystals found in Lake Ørn, Denmark, (O'Connell et al., 2015) and the deepest part of the Bothnian Sea (site US5B; Egger et al., 2015a; Fig. 7a) at all sieved intervals below the SMTZ (34–36, 49–52 and 55–58 cm). Quantitative analysis of the crystals with electron microprobe EDS revealed an average Fe : P ratio of 3.3 mol mol−1 and the presence of significant amounts of Mn, Mg, Si and Al (Fig. 7b; Table S5). In samples analyzed above and within the SMTZ such crystals were not observed.
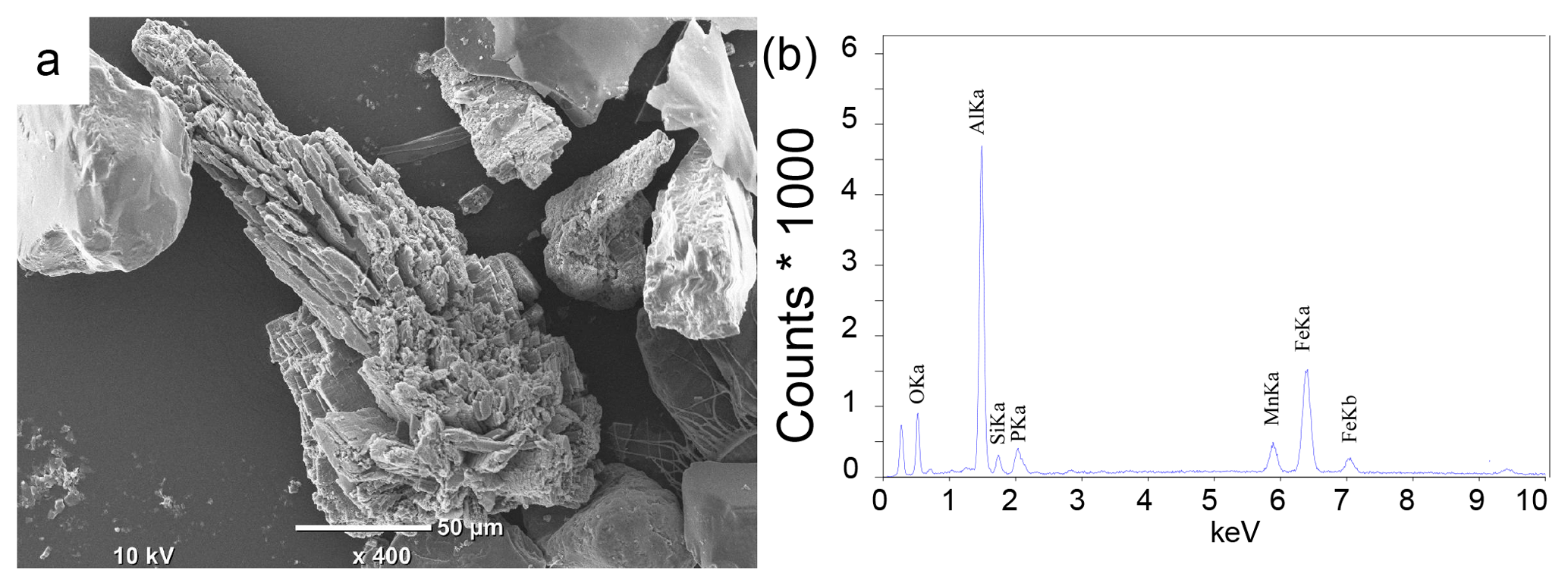
Figure 7(a) Scanning electron micrograph: example of a vivianite crystal from the 34–36 cm depth interval at site NB8 sampled in August 2015. (b) Electron microprobe EDS spectrum of a spot measurement on the surface of the vivianite crystal.
4.4 Model results
Modeled porewater and solid-phase depth profiles for site NB8 capture the main trends in the measured profiles (Figs. 4, 5 and 6). We note that the model slightly underestimates the amount of Fe-bound P in the upper 15 cm of the sediment and overestimates organic P. The observed variations in total S, total Mn and Fe-P below 30 cm depth in the sediment were not targeted with the model.
Depth integrated rates of P cycling in 2015 (Fig. 8) show that the release of from organic P accounts for only 2.3 % of the release to the porewater at site NB8. The main source of is the release from Fe oxides, with H2S-driven reductive dissolution accounting for 61 % of the release and dissolution coupled to organic matter degradation accounting for 28 % (Fig. 8). In the model, most of the Fe oxide reduction involves the less reactive beta fraction and takes place in the upper 20 cm of the sediment (Fig. S6; Reactions R14 and R15).
Approximately 30 % of the removal from the porewater takes place through release to the overlying water by bioirrigation and diffusion, with the former being the most important process. Binding of to Fe oxides accounts for 60 % of the removal while 10 % is precipitated as vivianite (Fig. 8). However, Fe oxides that are formed in situ are not a permanent sink for as the majority is dissolved through reductive dissolution. Burial of P as vivianite accounts for 41 % of the total P burial at site NB8 (Fig. 8). At depth, P bound to vivianite becomes more important compared to P bound to Fe oxides (Fig. S7).
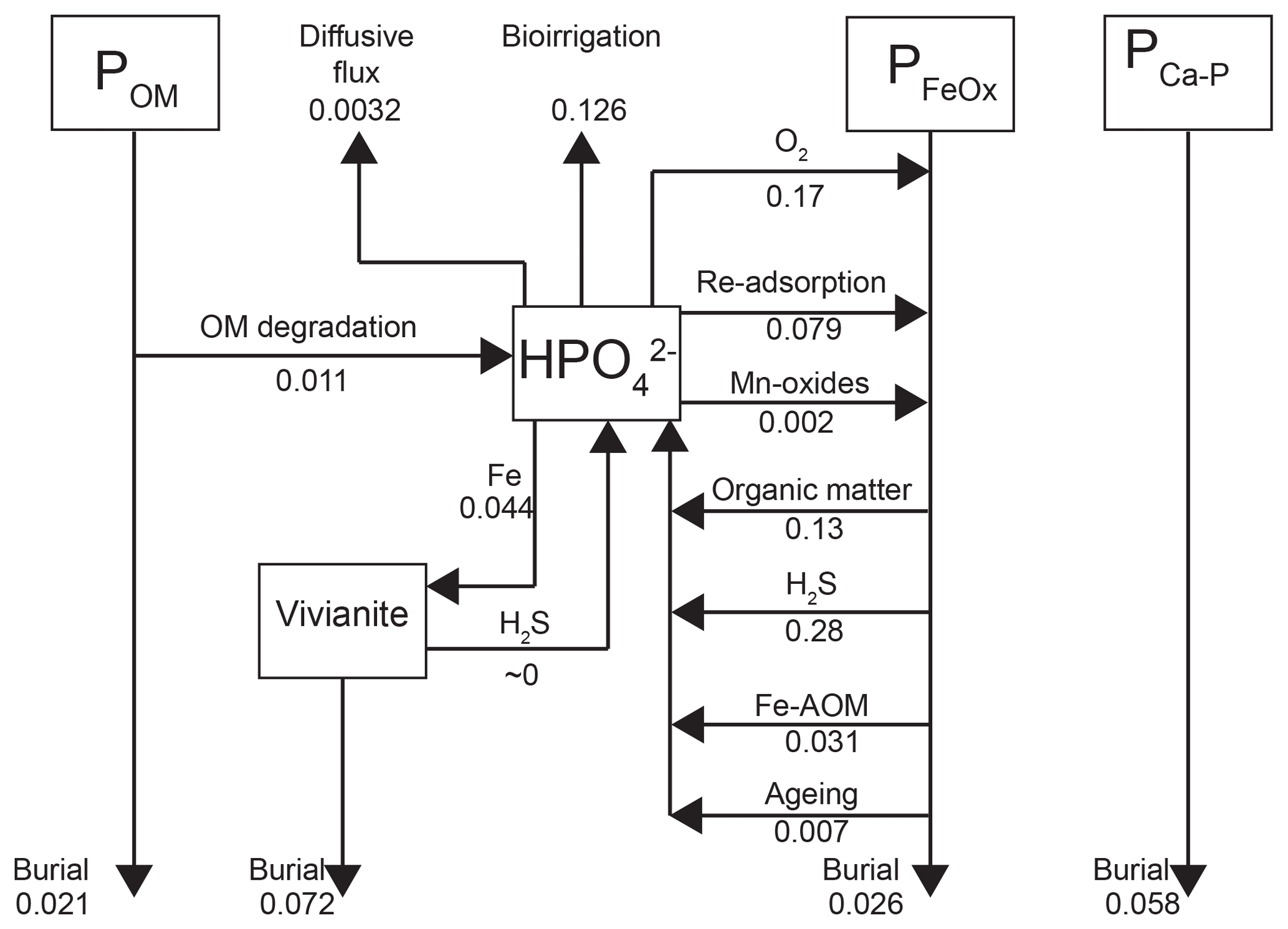
Figure 8Mass balance of P as calculated with the transient reactive transport model for 2015 (Fig. 3). Results are in non-steady state. Fluxes are in .
In the model, total P burial rates increased from 0.12 to 0.34 upon a decrease in salinity from 20 to 0 (Fig. 9). Changes in salinity did not affect the burial rates of Ca-P, organic P and P associated with Fe oxides. In the latter case, this was because only the unreactive fraction of Fe oxides remained. The percentage of P buried as vivianite increased strongly with decreasing salinity.
In the model scenario in which changes in the rates of sedimentation and organic matter and Fe oxide input were coupled, the total P burial rate increased from 0.025 to 0.45 when assuming sedimentation rates between 0.25 and 2 cm yr−1 (Fig. 9). At a sedimentation rate of 0.5 cm yr−1, little P was buried in the form of vivianite. At higher sedimentation rates, formation of vivianite in the sediment increased and the total burial of P was enhanced. Model runs in which changes in the same factors were evaluated separately revealed that increasing the rate of sedimentation or Fe oxide input both enhance vivianite formation and burial of P (Fig. 10a and b). The response to an increase in the rate of sedimentation is nonlinear, with the greatest increase in P burial between 0.5 and 0.85 cm yr−1. For organic matter, the model runs reveal an optimum input rate beyond which P and vivianite burial rates decrease (Fig. 10c).

Figure 9Sensitivity of (a) total phosphorus burial rate and (b) the burial forms of P at site NB8 to changes in salinity, as well as in sedimentation rate and inputs of organic matter and Fe oxides as calculated in the model. Four burial forms of P are distinguished: authigenic and detrital apatite (Ca-P), organic P (Org-P), P bound to Fe oxides (FeOx-P) and vivianite. The dashed line indicates the total P burial as a function of sedimentation rate in the coastal zone of the Baltic Sea as suggested in Asmala et al. (2017). Red stars are the burial rates of P as measured at our five sites (Table 1). Sedimentation rates are initial sedimentation rates at t=0.
5.1 Phosphorus burial in the Öre Estuary
Sediments in the coastal zone of the Bothnian Sea have been suggested to act as an efficient sink for P from rivers and the open sea based on budget calculations and an assumed average burial rate of P of 0.007 (Asmala et al., 2017). We find substantially higher rates of P burial in the Öre Estuary ranging from 0.026 to 0.145 (Table 1). If our data are representative for the wider area, sediments in the coastal zone of the northern Baltic Sea may be an even more efficient sink for P than previously thought.
In their study, Asmala et al. (2017) identified a linear relationship between P burial and rates of sedimentation for a range of coastal systems around the Baltic Sea, but excluding the northern areas because of a lack of data (Fig. 9). Strikingly, our study sites in the Öre Estuary are characterized by a higher burial of P than predicted by this relationship. This particularly holds for site NB8. In the following, we will assess whether vivianite formation can play a role in explaining this enhanced P retention. As discussed in detail by Egger et al. (2015a), vivianite formation is generally most pronounced in sediments below the SMTZ, where both Fe2+ and accumulate in the porewater due to dissolution of Fe oxides and release of associated P. While we find both solutes in the porewater at depth at all sites, the highest concentrations are observed at sites N7 and NB8 where a distinct SMTZ was present between ca. 10 and 20 cm depth in the sediment. The position of this SMTZ shows little seasonality, likely because of the refractory nature of the sediment organic matter, which is mostly of terrestrial origin, and the relatively limited seasonal change in bottom water temperature. This implies that any vivianite formed below the SMTZ will likely be preserved and buried, because there will be no further exposure to sulfide.
Detailed analyses of the sediments at site NB8 with SEM-EDS confirm the presence of vivianite crystals below the SMTZ and a lack thereof in and above the SMTZ (Fig. 7). This trend with depth points towards an authigenic origin. The average Fe : P ratio of the crystals of 3.3 mol mol−1 (Table S5) is higher than the 2 : 1 stoichiometric ratio of vivianite. Similar high ratios were also observed for vivianite crystals in sediments of the Landsort Deep in the Baltic Proper, where they were explained by surface enrichments of Fe (Dijkstra et al., 2016). The vivianite was also enriched in Mn and Mg, which are both elements that are known to be included in the structure of the mineral (Dijkstra et al., 2016, 2018; Egger et al., 2015a). The presence of Al and Si (Table S5) likely reflects the presence of clay particles that were not removed prior to the SEM-EDS analysis (Dijkstra et al., 2018; Egger et al., 2015a).
The Fe-P below the SMTZ (Fig. 6) is likely a mixture of both Fe-oxide-bound P and vivianite, as shown previously for sediments in the deepest part of the Bothnian Sea (Egger et al., 2015a). Due to dissolution of Fe oxides and vivianite formation in the sediment, the fraction of P bound to vivianite increases with depth (Fig. S7). The results of the reactive transport model suggest that Fe-oxide-bound P and vivianite account for ca. 15 % and 40 % of the total burial of P, respectively. Authigenic Ca-P and organic P concentrations are relatively low and each account for ca. 15 % of total P burial (Fig. S5). The remainder of the P is buried as nonreactive P.
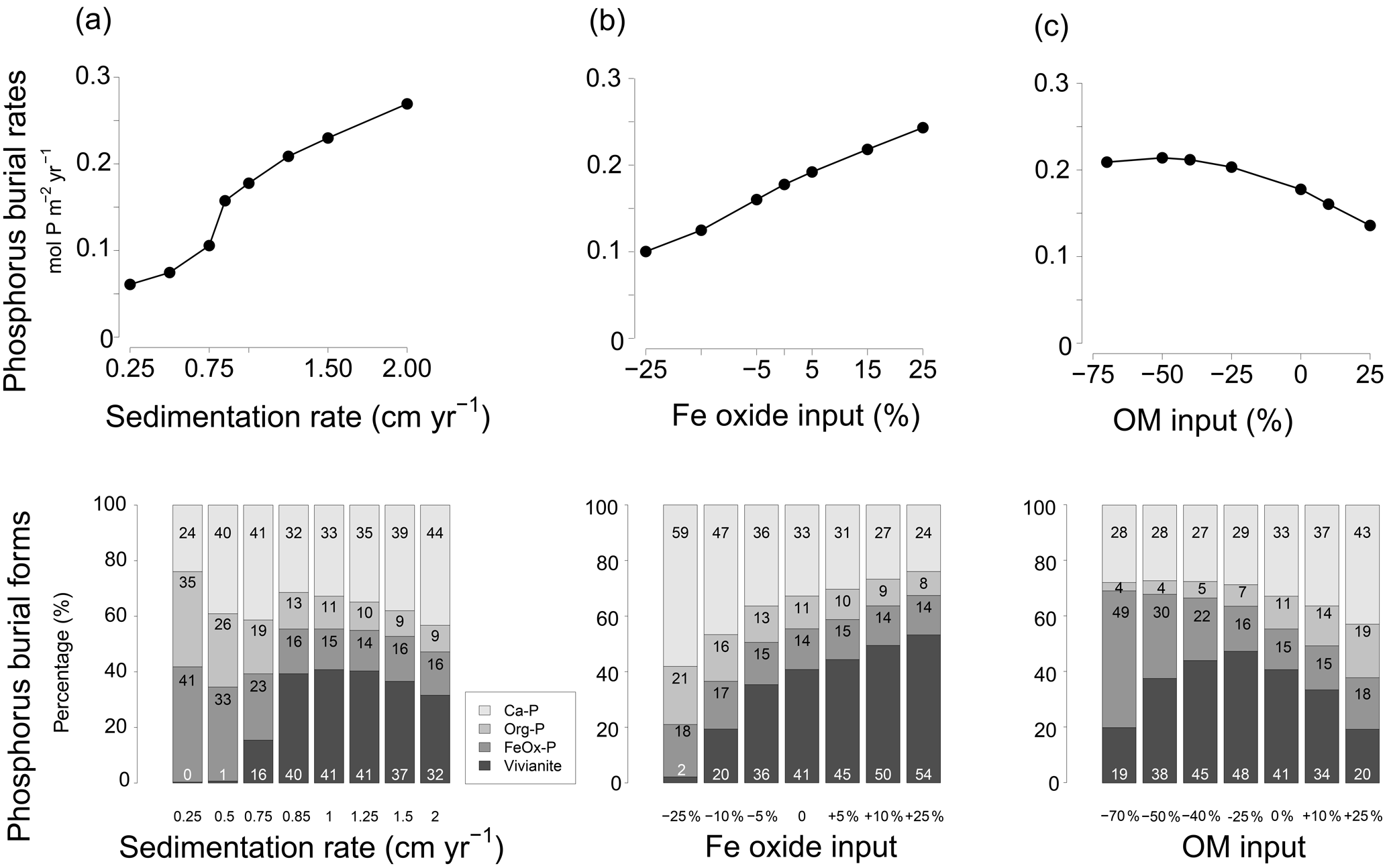
Figure 10Sensitivity analysis of P burial as a function of (a) changing sedimentation rate between 0.25 and 2 cm yr−1 and burial forms of P; (b) changing input of Fe oxides ranging between −25 % and 25 % and burial forms of P; (c) organic matter (OM) input ranging between −70 % and 25 % and burial forms of P.
5.2 Vivianite formation in coastal sediments in the northern Baltic Sea
Vivianite formation in sediments strongly depends on the balance between the formation of H2S and the input of Fe oxides (Ruttenberg, 2003). When there is an excess of Fe oxides over H2S, not all Fe oxides will be converted to Fe sulfides, and more Fe2+ will be available to precipitate with as vivianite (Gächter and Müller, 2003; Hartzell et al., 2017; Rozan et al., 2002). The results of the sensitivity analysis (Figs. 9 and 10) show that the bottom water salinity and input of Fe oxides play a critical role in controlling this balance, with vivianite formation and total P burial increasing strongly at lower salinities and with high Fe oxide inputs. A higher sedimentation rate also enhances the formation of vivianite and P burial because of more rapid burial of Fe oxides below the SMTZ. In our model scenario, sedimentation rates greater than 0.5 cm yr−1 were particularly conducive to vivianite formation. Vivianite formation is also highly sensitive to the input of organic matter. When the input of organic matter is lowered to −75 % of that assumed in the baseline scenario, the release of Fe2+ to the porewater due to Fe oxide reduction decreases to such an extent that formation of vivianite is limited by the availability of Fe2+. Consequently, P burial decreases. An increased input of organic matter relative to the baseline scenario enhances the formation of H2S and leads to a decline in vivianite formation and P burial. When assuming that changes in the rate of sedimentation and the input of Fe oxides and organic matter are coupled (Fig. 9), the net effect is an increase in vivianite formation and P burial. In summary, conditions for vivianite formation are most favorable in sediments with a low bottom water salinity, a high sedimentation rate and a high input of Fe oxides. While high rates of organic matter input are also required, there is an optimum rate above which vivianite formation declines.
5.3 Variations in riverine Fe and organic matter fluxes
In the Bothnian Sea, boreal rivers are the main source of Fe and organic matter to the coastal zone (Algesten et al., 2006; Björkvald et al., 2008; Palviainen et al., 2015). The input from rivers can be highly variable between years (Hölemann et al., 2005; Rember and Trefry, 2004; Sarkkola et al., 2013). The variations in sediment Fe, S and Fe/Al with depth, with maxima at depths of 21, 42 and 60 cm at site NB8, are in accordance with such a variation in input (Fig. 5). This is confirmed by the model, since a strong increase in input of Fe oxides and organic matter is required to describe the maxima in Fe and S in the SMTZ (Fig. 5) and the maximum in vivianite directly below it (Fig. 6). This increased input of Fe oxides and organic matter leads to enhanced conversion of Fe oxides to Fe sulfides. We suggest that these high inputs of Fe and organic matter are directly linked to variations in river discharge, that may be linked to negative phases in the North Atlantic Oscillation (Gillibrand et al., 2005). In 1996 a dry period affected the entire Bothnian Sea region (Fig. S8; Marttila et al., 2016). In that year, the Öre River discharge was unusually low (Fig. 2). In the following years, i.e., starting in 1997, this then led to an increased input of Fe downstream (Sarkkola et al., 2013). This timing coincides with the maximum in Fe and organic matter input assumed in our model scenario (Fig. 3). In the modeling study for the deep basin site in the Bothnian Sea the timing of the enhanced Fe input also corresponds to the year 1997 (Rooze et al., 2016), implying that a wide area was affected. Although not included in the model, a similar scenario would explain the enrichments of Fe, S and Fe-bound P at greater depth in the sediment, with the 1976 dry period leading to enhanced input of Fe and organic matter starting in 1977 and the Fe enrichment at 42 cm depth. In contrast to Fe, limited reoxidation of Mn is expected during a dry period because of less formation of reduced Mn species (Burdige, 1993). Therefore, no strong increase in riverine Mn input is expected after a dry period. In summary, our results show that variations in rainfall on land play a critical role in the transport of Fe and organic matter in rivers to the coastal zone. This riverine transport ultimately determines how much P is buried in the coastal zone. In the coastal Bothnian Sea, P burial is twice as high as the input of P from land, implying that large amounts of the buried P are imported from the open sea (Asmala et al., 2017). Part of the P in the open sea is known to be imported from the adjacent eutrophic Baltic Proper (Savchuk, 2005). The high P burial in the coastal zone coupled to the input of Fe from rivers likely contributes to the seasonal P limitation observed in the Bothnian Sea (Tamminen and Andersen, 2007).
5.4 Implications
In most marine systems, the balance between H2S formation and Fe oxide input is such that most Fe oxides are converted to Fe sulfides, thereby allowing only limited vivianite formation (Ruttenberg, 2003). Here we show that in estuaries with high inputs of Fe oxides from rivers, high sedimentation rates and sufficient input of organic matter this balance can become very favorable for the formation of vivianite. Such estuaries can act as a highly effective “coastal filter” for P from rivers and for P imported from the open sea (Asmala et al., 2017; Bouwman et al., 2013).
Our study also highlights the role of bottom water salinity in vivianite formation. While vivianite is a key sink for P in the Öre Estuary at a salinity of 5, we show that its formation is enhanced further at low salinity. Many studies focus only on the relatively high salinity parts of estuaries (e.g., as compiled for the Baltic Sea in Asmala et al., 2017). This may explain why the role of vivianite as a sink for P in these systems has been largely overlooked.
Sea level rise is expected to increase bottom water salinity in coastal areas in future (Scavia et al., 2002). In the Baltic Sea, however, salinity is expected to decrease because of enhanced precipitation on land and associated higher river runoff (Graham, 2004; Meier et al., 2006). Recent studies also indicate that Fe2+ input from rivers in Europe and North America increased over the past few decades, possibly due to changes in land use and expanded forestry (Björnerås et al., 2017; Kritzberg and Ekström, 2012). For the coastal zone of the Baltic Sea, both a decline in salinity and a continued enhanced input of Fe from rivers will promote conditions for vivianite formation in the sediment and increase its role as a sink for P. Detailed studies of other coastal areas are required to allow a prediction of the role of vivianite formation in the global coastal ocean, since an increased salinity and increased Fe input have opposite effects on the formation of vivianite.
Our study reveals a very high burial of phosphorus (P) in sediments of an oligotrophic estuary in the northern Bothnian Sea (up to 0.145 ). These sediments act as a highly efficient sink for P from rivers and the open sea. We demonstrate that the high P retention in the sediment is related to the formation of vivianite, which is visible in the form of discrete crystals below the sulfate methane transition zone at the site with the highest sedimentation rate. By combining P extractions and reactive transport modeling of solid-phase and porewater profiles, we demonstrate that vivianite accounts for 40 % of total P burial at the same site. With the model, we also assess the sensitivity of vivianite formation to the key drivers of variations in the availability of H2S versus Fe oxides. We show that vivianite formation is promoted when bottom water salinity is low and rates of sedimentation and inputs of Fe oxides are high. High organic matter inputs are also a requirement, but in our scenario there is a distinct optimum for vivianite formation.
We suggest that enrichments of Fe, S and P in the sediment are linked to periods of enhanced riverine input of Fe and organic matter. Two sets of enrichments can be coupled to the years 1977 and 1997, when riverine fluxes of Fe were likely enhanced in a period following a dry year on land in 1976 and 1996, respectively. The enhanced input of Fe and organic matter likely increased the formation of vivianite and the burial of P in the sediment.
In the future, climate change is expected to enhance the freshwater input to the Baltic Sea and thereby decrease its salinity. Continued elevated Fe2+ input from rivers is also expected. Both a decreasing salinity and an increased Fe input will create more favorable conditions for vivianite formation in the sediment. We therefore expect that, in future, vivianite will become more important as a sink for P in the coastal zone of the Baltic Sea.
The model code is available from Wytze K. Lenstra and Caroline P. Slomp upon request.
All data will be made available in the PANGAEA database. In the meantime data is available upon request to the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/bg-15-6979-2018-supplement.
CS, WL, NH and DC designed the research. WL, ME, NH and DC performed the analyses. WL performed the model simulations. WL, ME, NH, EK and CS interpreted the data. WL and CS wrote the paper with comments provided by ME, NH, EK and DC.
The authors declare that they have no conflict of interest.
We thank the captain, crew and scientific participants for
their assistance during sampling aboard R/V Lotty in April and
August 2015 and Dineke van de Meent, Thom Claessen, Ton Zalm, Arnold van Dijk,
Dominika Kasjaniuk and Martijn Hermans for analytical assistance. We
thank Christophe Rabouille for the analysis of porewater . We thank Mathilde Hagens for
assistance during reactive transport modeling. This research was funded by
the Netherlands Organisation for Scientific Research (NWO-Vici grant
865.13.005; to Caroline P. Slomp), ERC starting grant no. 278364 and this
study was partly supported by the BONUS COCOA project (grant agreement
2112932-1), funded jointly by the EU and FORMAS. The Swedish Meteorological
and Hydrological Institute (SMHI) is acknowledged for open source data of
macrofaunal abundance and river discharge.
Edited by: Clare Woulds
Reviewed by: two anonymous referees
Algesten, G., Brydsten, L., Jonsson, P., Kortelainen, P., Löfgren, S., Rahm, L., Räike, A., Sobek, S., Tranvik, L., Wikner, J., and Jansson, M.: Organic carbon budget for the Gulf of Bothnia, J. Marine Syst., 63, 155–161, https://doi.org/10.1016/j.jmarsys.2006.06.004, 2006. a, b, c, d, e
APHA: Standard methods for the examination of water and wastewater, 11th edn., American Journal of Public Health and the Nations Health, 51, 940, https://doi.org/10.2105/AJPH.51.6.940-a, 2005. a
Asmala, E., Carstensen, J., Conley, D., Slomp, C. P., Stadmark, J., and Voss, M.: Efficiency of the coastal filter: nitrogen and phosphorous removal in the Baltic Sea, Limnol. Oceanogr., 62, S222–S238, https://doi.org/10.1002/lno.10644, 2017. a, b, c, d, e, f, g, h
Bartlett, J. K. and Skoog, D. A.: Colorimetric determination of elemental sulfur in hydrocarbons, Anal. Chem., 26, 1008–1011, https://doi.org/10.1021/ac60090a014, 1954. a
Beal, E. J., House, C. H., and Orphan, V. J.: Manganese- and iron-dependent marine methane oxidation, Science, 325, 184–187, https://doi.org/10.1126/science.1169984, 2009. a
Björkvald, L., Buffam, I., Laudon, H., and Mörth, C. M.: Hydrogeochemistry of Fe and Mn in small boreal streams: The role of seasonality, landscape type and scale, Geochim. Cosmochim. Ac., 72, 2789–2804, https://doi.org/10.1016/j.gca.2008.03.024, 2008. a, b, c, d, e
Björnerås, C., Weyhenmeyer, G. A., Evans, C. D., Gessner, M. O., Grossart, H.-P., Kangur, K., Kokorite, I., Kortelainen, P., Laudon, H., Lehtoranta, J., Lottig, N., Monteith, D. T., Nõges, P., Nõges, T., Oulehle, F., Riise, G., Rusak, J. A., Räike, A., Sire, J., Serling, S., and Kritzberg, E. S.: Widespread increases in iron concentration in European and North American freshwaters, Global Biogeochem. Cy., 31, 1488–1500, https://doi.org/10.1002/2017GB005749, 2017. a, b
Boudreau, B. P.: On the equivalence of nonlocal and radial-diffusion models for porewater irrigation, J. Mar. Res., 42, 731–735, 1984. a
Boudreau, B. P.: Diagenetic models and their implementation – Modelling transport and reactions in aquatic sediments, ISBN-13:978-3-642-64399-6, 1997. a
Bouwman, A. F., Bierkens, M. F. P., Griffioen, J., Hefting, M. M., Middelburg, J. J., Middelkoop, H., and Slomp, C. P.: Nutrient dynamics, transfer and retention along the aquatic continuum from land to ocean: towards integration of ecological and biogeochemical models, Biogeosciences, 10, 1–22, https://doi.org/10.5194/bg-10-1-2013, 2013. a, b
Burdige: The biogeochemistry of manganese and iron reduction in marine sediments, Earth Sci. Rev., 35, 249–284, https://doi.org/10.1016/0012-8252(93)90040-E, 1993. a
Burdige, D. J.: Geochemistry of Marine Sediments, Princeton University Press, Princeton, 609 pp., https://doi.org/10.1086/533614, 2006. a, b
Burton, E. D., Bush, R. T., and Sullivan, L. A.: Fractionation and extractability of sulfur, iron and trace elements in sulfidic sediments, Chemosphere, 64, 1421–1428, https://doi.org/10.1016/j.chemosphere.2005.12.003, 2006. a
Burton, E. D., Sullivan, L. A., Bush, R. T., Johnston, S. G., and Keene, A. F.: A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils, Appl. Geochem., 23, 2759–2766, https://doi.org/10.1016/j.apgeochem.2008.07.007, 2008. a
Cline, J. D.: Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 14, 454–458, https://doi.org/10.4319/lo.1969.14.3.0454, 1969. a
Delaney, M. L.: Phosphorus accumulation in marine sediments and the oceanic phosphorus cycle, Global Biogeochem. Cy., 12, 563–572, https://doi.org/10.1029/98GB02263, 1998. a
Dijkstra, N., Kraal, P., Kuypers, M. M. M., Schnetger, B., and Slomp, C. P.: Are iron-phosphate minerals a sink for phosphorus in anoxic Black Sea sediments?, PLoS ONE, 9, 1–12, https://doi.org/10.1371/journal.pone.0101139, 2014. a
Dijkstra, N., Slomp, C. P., and Behrends, T.: Vivianite is a key sink for phosphorus in sediments of the Landsort Deep, an intermittently anoxic deep bsin in the Baltic Sea, Chem. Geol., 438, 58–72, https://doi.org/10.1016/j.chemgeo.2016.05.025, 2016. a, b, c, d
Dijkstra, N., Hagens, M., Egger, M., and Slomp, C. P.: Post-depositional formation of vivianite-type minerals alters sediment phosphorus records, Biogeosciences, 15, 861–883, https://doi.org/10.5194/bg-15-861-2018, 2018. a, b
Egger, M., Jilbert, T., Behrends, T., Rivard, C., and Slomp, C. P.: Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments, Geochim. Cosmochim. Ac., 169, 217–235, https://doi.org/10.1016/j.gca.2015.09.012, 2015a. a, b, c, d, e, f, g, h, i, j, k, l
Egger, M., Rasigraf, O., Sapart, C. J., Jilbert, T., Jetten, M. S. M., Röckmann, T., Van Der Veen, C., Bânda, N., Kartal, B., Ettwig, K. F., and Slomp, C. P.: Iron-mediated anaerobic oxidation of methane in brackish coastal sediments, Environ. Sci. Technol., 49, 277–283, https://doi.org/10.1021/es503663z, 2015b. a, b
Emerson, S., Jahnke, R., and Heggie, D.: Sediment-water exchange in shallow water estuarine sediments, J. Marine Res., 42, 709–730, https://doi.org/10.1357/002224084788505942, 1984. a
Ettwig, K. F., Zhu, B., Speth, D., Keltjens, J. T., Jetten, M. S. M., and Kartal, B.: Archaea catalyze iron-dependent anaerobic oxidation of methane, P. Natl. Acad. Sci. USA, 113, 12792–12796, https://doi.org/10.1073/pnas.1609534113, 2016. a
Fleming-Lehtinen, V. and Laamanen, M.: Long-term changes in Secchi depth and the role of phytoplankton in explaining light attenuation in the Baltic Sea, Estuar. Coast. Shelf S., 102–103, 1–10, https://doi.org/10.1016/j.ecss.2012.02.015, 2012. a
Fleming-Lehtinen, V., Laamanen, M., Kuosa, H., Haahti, H., and Olsonen, R.: Long-term development of inorganic nutrients and chlorophyll alpha in the open northern Baltic Sea, Ambio, 37, 86–92, https://doi.org/10.1579/0044-7447(2008)37[86:LDOINA]2.0.CO;2, 2008. a
Froelich, P. N.: Kinetic control of dissolved phosphate in natural rivers and estuaries: A primer on the phosphate buffer mechanism, Limnol. Oceanogr., 33, 649–668, https://doi.org/10.2307/2837216, 1988. a
Froelich, P. N., Bender, M. L., and Luedtke, N. A.: The marine phosphorus cycle, 4, 105, https://doi.org/10.3389/fmicb.2013.00105, 1982. a
Froelich, P. N., Arthur, M. A., Burnett, W. C., Deakin, M., Hensley, V., Jahnke, R., Kaul, L., Kim, K. H., Roe, K., Soutar, A., and Vathakanon, C.: Early diagenesis of organic matter in Peru continental margin sediments: Phosphorite precipitation, Mar. Geol., 80, 309–343, https://doi.org/10.1016/0025-3227(88)90095-3, 1988. a
Gächter, R. and Müller, B.: Why the phosphorus retention of lakes does not necessarily depend on the oxygen supply to their sediment surface, Limnol. Oceanogr., 48, 929–933, https://doi.org/10.4319/lo.2003.48.2.0929, 2003. a
Gillibrand, P. A., Cage, A. G., and Austin, W. E.: A preliminary investigation of basin water response to climate forcing in a Scottish fjord: Evaluating the influence of the NAO, Cont. Shelf Res., 25, 571–587, https://doi.org/10.1016/j.csr.2004.10.011, 2005. a
Graham, L. P.: Climate change effects on river flow to the Baltic Sea, Ambio, 33, 235–241, https://doi.org/10.1639/0044-7447(2004)033, 2004. a
Gunnars, A., Blomqvist, S., Johansson, P., and Andersson, C.: Formation of Fe(III) oxyhydroxide colloids in freshwater and brackish seawater, with incorporation of phosphate and calcium, Geochim. Cosmochim. Ac., 66, 745–758, https://doi.org/10.1016/S0016-7037(01)00818-3, 2002. a
Harley, C. D. G., Hughes, A. R., Hultgren, K. M., Miner, B. G., Sorte, C. J. B., Thornber, C. S., Rodriguez, L. F., Tomanek, L., and Williams, S. L.: The impacts of climate change in coastal marine systems, Ecol. Lett., 9, 228–241, https://doi.org/10.1111/j.1461-0248.2005.00871.x, 2006. a
Hartzell, J. L., Jordan, T. E., and Cornwell, J. C.: Phosphorus sequestration in sediments along the salinity gradients of Chesapeake Bay subestuaries, Estuaries and Coasts, 40, 1607–1625, 2017. a, b
Hellemann, D., Tallberg, P., Bartl, I., Voss, M., and Hietanen, S.: Denitrification in an oligotrophic estuary: A delayed sink for riverine nitrate, Mar. Ecol. Prog. Ser., 583, 63–80, https://doi.org/10.3354/meps12359, 2017. a
Hölemann, J. A., Schirmacher, M., and Prange, A.: Seasonal variability of trace metals in the Lena River and the southeastern Laptev Sea: Impact of the spring freshet, Global Planet. Change, 48, 112–125, https://doi.org/10.1016/j.gloplacha.2004.12.008, 2005. a, b
Hsu, T.-W., Jiang, W.-T., and Wang, Y.: Authigenesis of vivianite as influenced by methane-induced sulfidization in cold-seep sediments off southwestern Taiwan, J. Asian Earth Sci., 89, 88–97, https://doi.org/10.1016/j.jseaes.2014.03.027, 2014. a
Jordan, T. E., Cornwell, J. C., Boynton, W. R., and Anderson, J. T.: Changes in phosphorus biogeochemistry along an estuarine salinity gradient: The iron conveyer belt, Limnol. Oceanogr., 53, 172–184, https://doi.org/10.4319/lo.2008.53.1.0172, 2008. a
Jørgensen, B. B.: A comparison of methods for the quantification of bacterial sulfate reduction in coastal marine sediments, Geomicrobiol. J., 1, 49–64, https://doi.org/10.1080/01490457809377723, 1978. a
Kauppi, L., Norkko, A., and Norkko, J.: Large-scale species invasion into a low-diversity system: spatial and temporal distribution of the invasive polychaetes Marenzelleria spp. in the Baltic Sea, Biol. Invasions, 17, 2055–2074, https://doi.org/10.1007/s10530-015-0860-0, 2015. a, b
Kraal, P. and Slomp, C. P.: Rapid and extensive alteration of phosphorus speciation during oxic storage of wet sediment samples, PLoS ONE, 9, e96859, https://doi.org/10.1371/journal.pone.0096859, 2014. a, b
Kraal, P., Slomp, C. P., Forster, A., Kuypers, M. M. M., and Sluijs, A.: Pyrite oxidation during sample storage determines phosphorus fractionation in carbonate-poor anoxic sediments, Geochim. Cosmochim. Ac., 73, 3277–3290, https://doi.org/10.1016/j.gca.2009.02.026, 2009. a, b
Kritzberg, E. S. and Ekström, S. M.: Increasing iron concentrations in surface waters – a factor behind brownification?, Biogeosciences, 9, 1465–1478, https://doi.org/10.5194/bg-9-1465-2012, 2012. a, b
Kuma, K., Nishioka, J., and Matsunaga, K.: Controls on iron(III) hydroxide solubility in seawater: The influence of pH and natural organic chelators, Limnol. Oceanogr., 41, 396–407, https://doi.org/10.4319/lo.1996.41.3.0396, 1996. a
Laudon, H., Berggren, M., Agren, A., Buffam, I., Bishop, K., Grabs, T., Jansson, M., and Kohler, S.: Patterns and Dynamics of Dissolved Organic Carbon (DOC) in Boreal Streams: The Role of Processes, Connectivity, and Scaling, Ecosystems, 14, 880–893, https://doi.org/10.1007/s10021-011-9452-8, 2011. a
Leipe, T., Tauber, F., Vallius, H., Virtasalo, J., Uścinowicz, S., Kowalski, N., Hille, S., Lindgren, S., and Myllyvirta, T.: Particulate organic carbon (POC) in surface sediments of the Baltic Sea, Geo-Mar. Lett., 31, 175–188, 2011. a
Li, W., Joshi, S. R., Hou, G., Burdige, D. J., Sparks, D. L., and Jaisi, D. P.: Characterizing Phosphorus Speciation of Chesapeake Bay Sediments Using Chemical Extraction, 31P NMR, and X-ray Absorption Fine Structure Spectroscopy, Environ. Sci. Technol., 49, 203–211, https://doi.org/10.1021/es504648d, 2015. a
Madison, A. S., Tebo, B. M., and Luther III, G. W.: Simultaneous determination of soluble manganese (III), manganese (II) and total manganese in natural (pore) waters, Talanta, 84, 374–381, 2011. a
Malmgren, L. and Brydsten, L.: Sedimentation of river-transported particles in the Ore estuary, northern Sweden, Hydrobiologia, 235–236, 59–69, https://doi.org/10.1007/BF00026200, 1992. a
Marttila, H., Nieminen, M. L., Arje, J., Meissner, K., Tuukkanen, T., Saukkoriipi, J., and Klove, B.: Dynamics and temporal changes in suspended sediment transport in northern Finland: transport of very fine particulate matter, Boreal Environ. Res., 21, 541–555, 2016. a
März, C., Hoffmann, J., Bleil, U., de Lange, G. J., and Kasten, S.: Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean), Mar. Geol., 255, 118–130, https://doi.org/10.1016/j.margeo.2008.05.013, 2008. a
Meier, H. E. M., Kjellström, E., and Graham, L. P.: Estimating uncertainties of projected Baltic Sea salinity in the late 21st century, Geophysical Research Letters, 33, 1–4, https://doi.org/10.1029/2006GL026488, 2006. a, b
Middelburg, J. J.: A simple rate model for organic matter decomposition in marine sediments, Geochim. Cosmochim. Ac., 53, 1577–1581, https://doi.org/10.1016/0016-7037(89)90239-1, 1989. a
Nembrini, G. P., Capobianco, J. A., Viel, M., and Williams, A. F.: A Mössbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy), Geochim. Cosmochim. Ac., 47, 1459–1464, https://doi.org/10.1016/0016-7037(83)90304-6, 1983. a
Norkko, J., Reed, D. C., Timmermann, K., Norkko, A., Gustafsson, B. G., Bonsdorff, E., Slomp, C. P., Carstensen, J., and Conley, D. J.: A welcome can of worms? Hypoxia mitigation by an invasive species, Glob. Change Biol., 18, 422–434, https://doi.org/10.1111/j.1365-2486.2011.02513.x, 2012. a
O'Connell, D. W., Mark Jensen, M., Jakobsen, R., Thamdrup, B., Joest Andersen, T., Kovacs, A., and Bruun Hansen, H. C.: Vivianite formation and its role in phosphorus retention in Lake Orn, Denmark, Chem. Geol., 409, 42–53, https://doi.org/10.1016/j.chemgeo.2015.05.002, 2015. a
Palviainen, M., Lehtoranta, J., Ekholm, P., Ruoho-Airola, T., and Kortelainen, P.: Land cover controls the export of terminal electron acceptors from boreal catchments, Ecosystems, 18, 343–358, https://doi.org/10.1007/s10021-014-9832-y, 2015. a
Pokrovsky, O. S. and Schott, J.: Iron colloids/organic matter associated transport of major and trace elements in small boreal rivers and their estuaries (NW Russia), Chem. Geol., 190, 141–179, https://doi.org/10.1016/S0009-2541(02)00115-8, 2002. a
Poulton, S. W. and Canfield, D. E.: Development of a sequential extraction procedure for iron: Implications for iron partitioning in continentally derived particulates, Chem. Geol., 214, 209–221, https://doi.org/10.1016/j.chemgeo.2004.09.003, 2005. a
Redfield, A. C.: The Biological Control of Chemical Factors in the Environment, Am. Sci., 46, 205–221, https://doi.org/10.2307/27828530, 1958. a
Reed, D. C., Gustafsson, B. G., and Slomp, C. P.: Shelf-to-basin iron shuttling enhances vivianite formation in deep Baltic Sea sediments, Earth Planet. Sci. Lett., 434, 241–251, https://doi.org/10.1016/j.epsl.2015.11.033, 2016. a
Rember, R. D. and Trefry, J. H.: Increased concentrations of dissolved trace metals and organic carbon during snowmelt in rivers of the Alaskan Arctic, Geochim. Cosmochim. Ac., 68, 477–489, https://doi.org/10.1016/S0016-7037(03)00458-7, 2004. a, b
Renz, J. R. and Forster, S.: Are similar worms different? A comparative tracer study on bioturbation in the three sibling species Marenzelleria arctia, M. viridis, and M. neglecta from the Baltic Sea, Limnol. Oceanogr., 58, 2046–2058, https://doi.org/10.4319/lo.2013.58.6.2046, 2013. a
Roden, E. E. and Edmonds, J. W.: Phosphate mobilization in iron-rich anaerobic sediments: Microbial Fe(III) oxide reduction versus iron-sulfide formation, Arch. Hydrobiol., 139, 347–378, 1997. a
Rooze, J., Egger, M., Tsandev, I., and Slomp, C. P.: Iron-dependent anaerobic oxidation of methane in coastal surface sediments: Potential controls and impact, Limnol. Oceanogr., 61, S267–S282, https://doi.org/10.1002/lno.10275, 2016. a, b, c, d, e, f, g
Rozan, T. F., Taillefert, M., Trouwborst, R. E., Glazer, B. T., Ma, S., Herszage, J., Valdes, L. M., Price, K. S., and Luther III, G. W.: Iron sulfur phosphorus cycling in the sediments of a shallow coastal bay: Implications for sediment nutrient release and benthic macroalgal blooms, Limnol. Oceanogr., 47, 1346–1354, https://doi.org/10.4319/lo.2002.47.5.1346, 2002. a
Rue, E. L. and Bruland, K. W.: Complexation of iron(III) by natural organic ligands in the Central North Pacific as determined by a new competitive ligand equilibration/adsorptive cathodic stripping voltammetric method, Mar. Chem., 50, 117–138, https://doi.org/10.1016/0304-4203(95)00031-L, 1995. a
Ruttenberg, K. C.: Development of a sequential extraction method for different forms of phosphorus in marine sediments, Limnol. Oceanogr., 37, 1460–1482, https://doi.org/10.4319/lo.1992.37.7.1460, 1992. a
Ruttenberg, K. C.: The Global Phosphorus Cycle, in: Treatise on Geochemistry, 2nd edn., 10, 499–558, https://doi.org/10.1016/B978-0-08-095975-7.00813-5, 2003. a, b, c, d, e, f, g
Ruttenberg, K. C. and Berner, R. A.: Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments, Geochim. Cosmochim. Ac., 57, 991–1007, https://doi.org/10.1016/0016-7037(93)90035-U, 1993. a, b
Sarkkola, S., Nieminen, M., Koivusalo, H., Lauren, A., Kortelainen, P., Mattsson, T., Palviainen, M., Piirainen, S., Starr, M., and Finer, L.: Iron concentrations are increasing in surface waters from forested headwater catchments in eastern Finland, Sci. Total Environ., 463–464, 683–689, https://doi.org/10.1016/j.scitotenv.2013.06.072, 2013. a, b, c, d, e
Savchuk, O. P.: Resolving the Baltic Sea into seven subbasins: N and P budgets for 1991–1999, J. Marine Syst., 56, 1–15, https://doi.org/10.1016/j.jmarsys.2004.08.005, 2005. a
Scavia, D., Field, J. C., Boesch, D. F., Buddemeier, R. W., Burkett, V., Cayan, D. R., Fogarty, M., Harwell, M. A., Howarth, R. W., Mason, C., Reed, D. J., Royer, T. C., Sallenger, A. H., and Titus, J. G.: Climate change impacts on U.S. coastal and marine ecosystems, Estuaries, 149, 149–164, https://doi.org/10.1007/BF02691304, 2002. a
Schlitzer, R.: Ocean Data View, 2012, available at: http://odv.awi.de (last access: 14 November 2018), 2015. a
Schwertmann, U. and Taylor, R. M.: Iron Oxides, in: Minerals in Soil Environments, 8, 379–438, https://doi.org/10.2136/sssabookser1.2ed.c8, 1989. a
Slomp, C.: Phosphorus cycling in the estuarine and coastal zones, in: Treatise on Estuarine and Coastal Science, 201–229, https://doi.org/10.1016/B978-0-12-374711-2.00506-4, 2011. a
Slomp, C., Van der Gaast, S., and Van Raaphorst, W.: Phosphorus binding by poorly crystalline iron oxides in North Sea sediments, Mar. Chem., 52, 55–73, https://doi.org/10.1016/0304-4203(95)00078-X, 1996a. a, b
Slomp, C. P., Epping, E. H. G., Helder, W., and Raaphorst, W. V.: A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments, J. Mar. Res., 54, 1179–1205, https://doi.org/10.1357/0022240963213745, 1996b. a
Slomp, C. P., Mort, H. P., Jilbert, T., Reed, D. C., Gustafsson, B. G., and Wolthers, M.: Coupled dynamics of iron and phosphorus in sediments of an oligotrophic coastal basin and the impact of anaerobic oxidation of methane, PLoS ONE, 8, e62386, https://doi.org/10.1371/journal.pone.0062386, 2013. a, b
Soetaert, K. and Herman, P. M. J.: A practical guide to ecological modelling: using R as a simulation platform, Springer Science & Business Media, 2008. a
Soetaert, K., Herman, P. M. J., and Middelburg, J. J.: A model of early diagenetic processes from the shelf to abyssal depths, Geochim. Cosmochim. Ac., 60, 1019–1040, https://doi.org/10.1016/0016-7037(96)00013-0, 1996. a
Solorzano, L.: Determination of ammonia in natural waters by the phenolhypochlorite method, Limnol. Oceanogr., 14, 799–801, https://doi.org/10.4319/lo.1969.14.5.0799, 1969. a
Stockenberg, A. and Johnstone, R. W.: Benthic Denitrification in the Gulf of Bothnia, Estuar. Coast. Shelf S., 45, 835–843, https://doi.org/10.1006/ecss.1997.0271, 1997. a
Strickland, J. D. H. and Parsons, T. R.: A Practical Handbook of Seawater Analysis, Fisheries Research Board of Canada, Ottawa, 55, 167, https://doi.org/10.1002/iroh.19700550118, 1972. a
Tamminen, T. and Andersen, T.: Seasonal phytoplankton nutrient limitation patterns as revealed by bioassays over Baltic Sea gradients of salinity and eutrophication, Mar. Ecol. Prog. Ser., 340, 121–138, https://doi.org/10.3354/meps340121, 2007. a
Van Cappellen, P. and Wang, Y.: Cycling of iron and manganese in surface sediments: A general theory for the coupled transport and reaction of carbon, oxygen, nitrogen, sulfur, iron, and manganese, Am. J. Sci., 296, 197–243, https://doi.org/10.2475/ajs.296.3.197, 1996. a
Van Santvoort, P. J. M., De Lange, G. J., Thomson, J., Colley, S., Meysman, F. J. R., and Slomp, C. P.: Oxidation and origin of organic matter in surficial Eastern Mediterranean hemipelagic sediments, Aquat. Geochem., 8, 153–175, https://doi.org/10.1023/A:1024271706896, 2002. a
Wang, Y. F. and Van Cappellen, P.: A multicomponent reactive transport model of early diagenesis: Application to redox cycling in coastal marine sediments, Geochim. Cosmochim. Ac., 60, 2993–3014, https://doi.org/10.1016/0016-7037(96)00140-8, 1996. a, b
Westrich, J. T. and Berner, R. A.: The role of sedimentary organic matter in bacterial sulfate reduction: The G model tested, Limnol. Oceanogr., 29, 236–249, https://doi.org/10.4319/lo.1984.29.2.0236, 1984. a
Wu, J. and Luther, G. W.: Complexation of Fe(III) by natural organic ligands in the Northwest Atlantic Ocean by a competitive ligand equilibration method and a kinetic approach, Mar. Chem., 50, 159–177, https://doi.org/10.1016/0304-4203(95)00033-N, 1995. a