the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 May 2019
| 13 May 2019
Sulfate deprivation triggers high methane production in a disturbed and rewetted coastal peatland
Franziska Koebsch
Matthias Winkel
Susanne Liebner
Julia Westphal
Iris Schmiedinger
Alejandro Spitzy
Matthias Gehre
Gerald Jurasinski
Stefan Köhler
Viktoria Unger
Marian Koch
Torsten Sachs
Michael E. Böttcher
In natural coastal wetlands, high supplies of marine sulfate suppress methanogenesis. Coastal wetlands are, however, often subject to disturbance by diking and drainage for agricultural use and can turn to potent methane sources when rewetted for remediation. This suggests that preceding land use measures can suspend the sulfate-related methane suppressing mechanisms. Here, we unravel the hydrological relocation and biogeochemical S and C transformation processes that induced high methane emissions in a disturbed and rewetted peatland despite former brackish impact. The underlying processes were investigated along a transect of increasing distance to the coastline using a combination of concentration patterns, stable isotope partitioning, and analysis of the microbial community structure. We found that diking and freshwater rewetting caused a distinct freshening and an efficient depletion of the brackish sulfate reservoir by dissimilatory sulfate reduction (DSR). Despite some legacy effects of brackish impact expressed as high amounts of sedimentary S and elevated electrical conductivities, contemporary metabolic processes operated mainly under sulfate-limited conditions. This opened up favorable conditions for the establishment of a prospering methanogenic community in the top 30–40 cm of peat, the structure and physiology of which resemble those of terrestrial organic-rich environments. Locally, high amounts of sulfate persisted in deeper peat layers through the inhibition of DSR, probably by competitive electron acceptors of terrestrial origin, for example Fe(III). However, as sulfate occurred only in peat layers below 30–40 cm, it did not interfere with high methane emissions on an ecosystem scale. Our results indicate that the climate effect of disturbed and remediated coastal wetlands cannot simply be derived by analogy with their natural counterparts. From a greenhouse gas perspective, the re-exposure of diked wetlands to natural coastal dynamics would literally open up the floodgates for a replenishment of the marine sulfate pool and therefore constitute an efficient measure to reduce methane emissions.





Coastal wetlands play an important role in climate change mitigation and adaption as they can efficiently accrete organic sediments, adjust coastal elevations to sea level rise and protect low-lying areas in the hinterland. Further, while freshwater wetlands constitute the largest natural source of the greenhouse gas methane (CH4, Zhang et al., 2017), the efficient accumulation of autochthonous C in coastal wetlands comes without the expense of high CH4 emissions (Holm et al., 2016). Methane is a potent greenhouse gas that is formed as a terminal product of organic matter breakdown under strictly anaerobic conditions typically in the absence of electron acceptors other than carbon dioxide (CO2) (Segers and Kengen, 1998). In coastal environments, methane production and emission are effectively suppressed by sulfate-rich seawaters: methanogens are outcompeted by sulfate-reducing bacteria (SRB) for acetate-type precursors and hydrogen (Schönheit et al., 1982; Lovley and Klug, 1983). This shifts the prevailing anaerobic C metabolic pathways from methanogenesis towards dissimilatory sulfate reduction (DSR) (Martens and Berner, 1974; King and Wiebe, 1980). In addition, sulfate () operates as an electron acceptor for anaerobic methane oxidation by a syntrophic consortium of anaerobic methanotrophs (ANMEs) and SRB (Iversen and Jorgensen, 1985; Boetius et al., 2000). Anaerobic methane oxidation has been specifically described for brackish wetland sediments but is not exclusively confined to the utilization of sulfate as an electron acceptor (Segarra et al., 2013, 2015).
Human activities such as diking and drainage place intensive pressure on coastal landscapes with sometimes irreversible impairments of their biogeochemical cycles and ecosystem functions (Karstens et al., 2016; Zhao et al., 2016). Dikes separate coastal wetlands from resupply of seawater, and drainage for agricultural use induces the aerobic decomposition of organic-rich sediments, resulting in substantial CO2 losses and land subsidence (Deverel and Rojstaczer, 1996; Miller, 2011; Deverel et al., 2016; Erkens et al., 2016). As sea levels are expected to rise, the controlled retreat from flood-prone areas becomes an essential strategy of integral coastal risk management to complement conventional technical solutions such as diking (Sánchez-Arcilla et al., 2016). Rewetting may re-establish the ability of abandoned coastal wetlands to efficiently accrete organic matter under anaerobic conditions and represents a promising management technique to reverse land surface subsidence caused by drainage-induced peat oxidation (Deverel et al., 2016; Erkens et al., 2016). Moreover, while freshwater wetlands may become methane sources upon rewetting (Wilson et al., 2009; Vanselow-Algan et al., 2015; Franz et al., 2016; Hemes et al., 2018), sulfate-rich seawater could potentially reduce post-rewetting methane release in coastal wetlands. However, recent work on a degraded brackish peatland has revealed high post-rewetting CH4 emissions (Hahn et al., 2015; Koebsch et al., 2015) and methanogen abundance (Wen et al., 2018), thereby challenging the common notion of coastal wetlands as negligible methane emitters. In fact, diking and the drainage-rewetting cycle may induce hydrological shifts and biogeochemical transformation processes that are so far not well understood. In particular, the transformation and/or relocation of the marine sulfate reservoir in the sediments of diked wetlands are of vital importance to understand the implications of anthropogenic intervention on coastal wetland biogeochemistry and to better constrain the climate effect of coastal wetland remediation.
Here, we investigate the mechanisms that allow for high methane production in disturbed and remediated coastal wetlands. We therefore address the fate of brackish compounds and the emerging S and C transformation processes in a rewetted, freshwater-fed peatland that was naturally exposed to episodic intrusions from the Baltic Sea. In the past, the peatland had been subject to intense human intervention including diking and drainage for agricultural use. After rewetting by freshwater-flooding, the site turned into a strong methane source. The underlying hydrological and biogeochemical processes were investigated along a brackish–terrestrial transect that spans between 300 and 1500 m in distance from the coastline using hydrogeochemical element patterns, stable isotope biogeochemistry, and microbiological analyses.
The specific goals were to
-
retrace the marine legacy effect remaining after diking and freshwater rewetting in the peat pore space using salinity, the isotope composition of water, and a suite of inert dissolved constituents that may be indicative for the intermingling of brackish and terrestrial waters.
-
track the fate of Baltic Sea-derived sulfate and uncover potential S transformation pathways using concentration patterns, stable isotope measurements of pore water (δ34S and δ18O), and solid S compounds as well as the bacterial community structure.
-
describe evolving methane cycling processes using concentration and stable isotope measurements of CH4 (δ13C, δ2H) and dissolved inorganic C (DIC, δ13C) as well as the abundance and community structure of methane-cycling microbes.
We hypothesized the marine legacy effect to be represented by a lateral gradient in electrical conductivity (EC) and pore water sulfate along the brackish–terrestrial transect. We further expected increasing terrestrial impact to promote the deprivation of the brackish sulfate pool and to induce complementary patterns of methane production.
2.1 Study site and sampling design
The study site is part of the nature reserve “Heiligensee und Hütelmoor”, a 490 ha coastal peatland complex located in NE Germany directly at the SW Baltic coast with an elevation between −0.3 and +0.7 m above sea level (Dahms, 1991) (latitude 54∘12′, longitude 12∘10′, Fig. 1). Climate is transitional maritime with continental influence from the east. The area receives a mean annual precipitation of 645 mm with a mean annual temperature of 9.2 ∘C (reference period 1982–2011, data from the German Weather Service, DWD). Peat formation was initiated by the Littorina Sea transgression and the postglacial sea level rise around 5400 BC. Presently, the Hütelmoor is fed by a 15 km2 forested catchment dominated by gley over fine sands. Originally, the fen exhibited 0.2–2.3 m deep layers of sulfidic reed-sedge peat underlain by Late Weichselian sands over impermeable till (Voigtländer et al., 1996; Bohne and Bohne, 2008). A total of 40 years of drainage for grassland use caused severe degradation of the peat, which was recently identified as sapric Histosol (Koebsch et al., 2013). Since the rewetting by flooding in 2010 through the construction of a weir at the outflow of the catchment, more than 80 % of the area has been permanently inundated with freshwater from the surrounding forest catchment (Miegel et al., 2016). Current vegetation of the Hütelmoor is dominated by patches of competitive emergent macrophytes such as reed and sedges (Phragmites australis (Cav.) Trin. ex Steud and Carex acutiformis Ehrh.) that increasingly supersede species indicative for brackish conditions (Bolboschoenus maritimus (L.) Palla, Schoenoplectus tabernaemontani (C. C. Gmel.) Palla) (Koch et al., 2017).
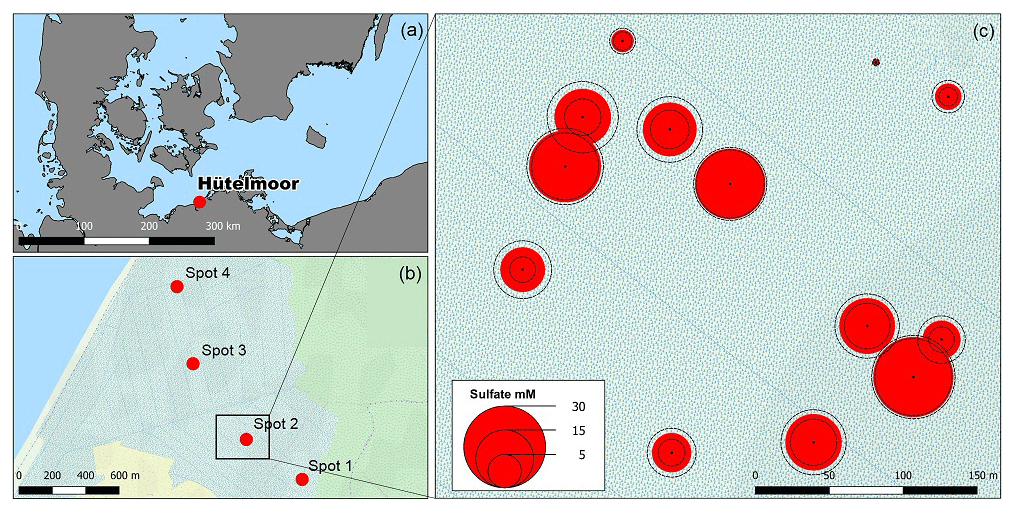
Figure 1(a) The study site Hütelmoor is located directly at the southwestern Baltic coast at an altitude between −0.2 and +0.2 m a.s.l. In its pristine state, the site was exposed to episodic brackish water intrusion by storm surges. (b) Profiles of sediments and pore waters were taken along a transect with 300–1500 m distance to the coastline. Deviations of the transect from the straight normal to the Baltic coastline arose due to the restricted accessibility of the site. (c) A former study located close to spot 2 in the center of the current sampling transect revealed high pore water sulfate concentrations in 30–60 cm below the surface with annual means up to 24±3 mM (red circles indicate annual means while dashed circle lines represent the standard deviation over the year). Map data are copyrighted under OpenStreetMap contributors and available from https://www.openstreetmap.org (last access: 23 November 2016).
Under natural coastal dynamics, the Hütelmoor is episodically flooded by storm surges. Low outflow and high evapotranspiration rates promote brackish conditions. Major brackish water intrusions were reported for 1904, 1913, 1949, 1954, and 1995 (Bohne and Bohne, 2008) though flooding frequency is reduced since the site was diked in 1903. Additional brackish input occurs through underground flow and atmospheric deposition as well as through high water situations at the Baltic Sea when backwater of the interconnected Warnow River delta enters the fen. However, potential brackish water entry paths other than storm surges have revealed a negligible effect on peat salinity (Selle et al., 2016). The last flooding event in 1995 raised EC in the drainage ditches up to 8 mS cm−1, but the EC decreased to the pre-flooding level of 2 mS cm−1 within the following 5 years (Bohne and Bohne, 2008).
Samples were collected at four spots along a transect with increasing distance to the Baltic Sea (300–1500 m, Fig. 1b) within 2 weeks in October/November 2014. The transect included the area of a former study which revealed high concentrations of brackish with annual means up to 23.7±3.2 mM (Koch, unpublished, Fig. 1c). At the time of sampling, water depth above the peat surface spanned from 9 to 19 cm, which presented the lowest range within the seasonal water level fluctuation. Sampling depth ranged from 45 to 65 cm, which was in most cases sufficient to cover the full peat depth including the underlying mineral soil.
2.2 Pore water analysis
Pore waters were collected from distinct depth below the surface (cm b.s.f.) with a stainless steel push-point sampler attached to a syringe to draw the sample from a distinct penetration depth. Temperature, pH, EC, and salinity were measured directly after sampling (Sentix 41 pH probe and a TetraCon 325 conductivity-measuring cell attached to a WTW multi 340i handheld; WTW, Weilheim). Samples were filtered (0.45 µm membrane syringe filters) in situ and transferred without headspace into vials (except for dissolved CH4). Vials had been previously preconditioned with 1 M HCl and subsequent 1 M NaOH and were filled with a compound-specific preservative (see below).
Dissolved CH4 concentration was measured with the headspace approach. Therefore, 5 mL of pore water was transferred into 12 mL septum-capped glass vials under atmospheric pressure. Before taking them to the field, the sampling vials were flushed with Ar and filled with 500 µL saturated HgCl solution to prevent further biological activity. After sampling, the punctuated septum was covered with lab foil and the vials were stored upside down to minimize CH4 loss. Headspace gas concentrations after equilibration were measured in duplicates with an Agilent 7890A gas chromatograph equipped with a flame ionization detector and with a carbon plot capillary column or HP-Plot Q (Porapak Q) column. Helium was used as the tracer gas. Gas sample analyses were performed after calibration of the gas chromatograph with gas standards (accuracy > 98.5 %). The measured headspace CH4 concentration was then converted into dissolved CH4 concentration using the temperature-corrected solubility coefficient (Wilhelm et al., 1977).
Samples for anion concentrations (, Cl−, Br−) were filled in 20 mL glass vials preserved with 1 mL 5 % ZnAc solution to prevent sulfide oxidation. Anion concentrations were analyzed by ion chromatography (Thermo Scientific Dionex) in a continuous flow of 9 mM NaCO3 eluent in an IonPac AS9-HC 4 column, partly after dilution of the sample. The device was calibrated with NIST standard reference material solutions freshly prepared before each run to span the concentration ranges of the (diluted) samples. Reproducibility between sample replicates was smaller than ±5 %.
For H2S analysis, pore water was filled into 5 mL polypropylene vials and preserved with 0.25 mL 5 % ZnAc solution. H2S concentration was measured photometrically (Specord 40, Analytic Jena) using the methylene blue method (Cline, 1969).
The metal and total dissolved S (TSdiss) concentrations were analyzed by ICP-OES (inductively coupled plasma optical emission spectrometry, iCAP 6300 DUO Thermo Fisher Scientific) after appropriate dilution. Since high amounts of dissolved organic carbon (DOC) may cause severe interferences in the ICP-OES element measurements, samples were boiled in Teflon beakers with 65 % HNO3 and subsequently 19 % HCl prior to analysis. The accuracy and precision were routinely checked with the certified CASS standards as described previously (Kowalski et al., 2012). The residual, non-specified S fraction (ResS resulting from the difference between TSdiss, H2S, and is suggested to consist primarily of dissolved organic S, polysulfides, and S intermediates.
δ13C and δD values of methane were analyzed using the gas chromatography–combustion technique (GC-C) and the gas chromatography–high-temperature-conversion technique (GC-HTC). The gas was directly injected in a gas chromatograph Agilent 7890 (Agilent Technologies, Germany), the peaks were separated using a CP-PoraBOND Q GC column (50 m × 0.32 mm × 5 µm, isotherm 60 ∘C, Varian). Methane was quantitatively converted to the analysis gases CO2 and H2 in the GC–Isolink interface (Thermo Finnigan, Germany) and directly transferred via open split interface (ConFlo IV, Thermo Finnigan, Germany). The δ13C and δD values of both gases were then measured with the isotope ratio mass spectrometer MAT 253 (Thermo Finnigan, Germany). Results for δ13C ratios of methane are given in the usual δ notation versus the Vienna PeeDee Belemnite (VPDB) standard. δD–CH4 ratios were referenced to the Vienna Standard Mean Ocean Water (V-SMOW).
The carbon isotope values (δ13C) of DIC were measured from a HgCl-preserved solution using a Thermo Finnigan MAT 253 gas mass spectrometer coupled to a Thermo Electron Gas Bench II via a Thermo Electron ConFlo IV split interface. NBS19 and LSVEC were used to scale the isotope measurements to the VPDB standard. Based on replicate measurements of standards, reproducibility was better than ±0.1 ‰ (Winde et al., 2014).
For the determination of sulfate isotope signatures, dissolved sulfate was precipitated with 5 % barium chloride as barium sulfate (Böttcher et al., 2007). After precipitation the solid was filtered, washed and dried, and further combusted in a Thermo Flash 2000 EA elemental analyzer that was connected to a Thermo Finnigan MAT 253 gas mass spectrometer via a Thermo Electron ConFlo IV split interface with a precision of better than ±0.2 ‰. Isotope ratios are converted to the Vienna Canyon Diablo Toilite (VCDT) scale (Mann et al., 2009). For oxygen isotope analyses, BaSO4 was decomposed by means of pyrolysis in silver cups using a high-temperature conversion elemental analyzer (HTO-, Hekatech, Germany) connected to an isotope gas mass spectrometer (Thermo Finnigan MAT 253) (Kornexl et al., 1999). The calibration took place via the reference materials IAEA-SO-5 and IAEA-SO-6 and 18O∕16O values were referenced to the V-SMOW standard. Replicate measurements agreed within ±0.5 ‰.
Stable oxygen (O) isotope measurements of pore waters were conducted using a CRDS system (Picarro L2140-i) versus the V-SMOW standard. International V-SMOW, SLAP, and GISP in addition to in-house standards were used to scale the isotope measurements.
2.3 Sediment analysis
Intact peat cores were collected with a perspex liner (ID: 59.5 mm) and subsequently punched out layer by layer. The peat section protruding from the end of the liner was divided into three subsamples for the analysis of (i) total reduced inorganic S (TRIS), (ii) total solid S (TSsolid) and reactive iron, and (iii) the microbial community structure. In order to minimize oxygen contamination, the outer layer of the peat core was omitted and subsamples were immediately packed. The aliquot for TRIS analysis was preserved with 1:1 (v∕v) 20 % ZnAc. Subsamples for microbial analysis were immediately stored in RNAlater stabilization solution to preserve DNA. A second core was taken for the analysis of water content and dry bulk density. TSsolid and TRIS samples were frozen within 8 h after collection. Aliquots for TSsolid elemental analysis were further freeze-dried and milled in a planet-ball mill.
TSsolid contents were analyzed by means of dry combustion using an Eltra CS-2000 after combustion at 1250 ∘C. The device was previously calibrated with a certified coal standard and precision is better than ±0.02 %.
TRIS fractions were determined by a two-step sequential extraction of iron monosulfides and pyrite (Fossing and Jørgensen, 1989). The acid volatile sulfur (AVS) fraction was extracted by the reaction with 1 M HCl for 1 h under a continuous stream of di-nitrogen gas. The H2S released was quantitatively precipitated as ZnS and then determined spectrophotometrically with a Specord 40 spectrophotometer following the method of Cline (1969). Chromium-reducible sulfur (CRS; essentially pyrite; FeS2), was extracted with hot acidic Cr(II)chloride solution. For δ34S analysis in different TRIS fractions the ZnS was converted to Ag2S by addition of 0.1 M AgNO3 solution with subsequent filtration, washing, and drying of the AgNO3 precipitate as described by Böttcher and Lepland (2000). The non-specified solid S fraction, resulting from the difference between TSsolid, CRS, and AVS, was suggested to present primarily organic-bond S (orgS). The δ34S composition of this residual fraction was measured from the washed and dried solid residue after the Cr(II) extraction step via C-IRmMS following the approach of Passier (1999). Reactive iron was extracted from freeze-dried sediments by the reaction with a 1 M HCl solution for 1 h (e.g., Canfield, 1989).
Iron was determined as Fe2+ after reduction with hydroxylamine hydrochloride via spectrophotometry using ferrozine as the complexing agent (Stookey, 1970). Reactive iron here is considered to be the sum of those iron fractions that may still react with dissolved sulfide. This fraction includes iron(III)oxyhydroxides and acid volatile sulfide (AVS, essentially FeS) as well as a very minor contribution from dissolved Fe2+ in the pore water (Canfield, 1989).
2.4 Microbial community analysis
Genomic DNA of 0.2–0.3 g of sediment was extracted with the EURx soil DNA kit (Roboklon, Berlin, Germany) according to manufactory protocols. DNA concentrations were quantified with a Nanophotometer® P360 (Implen GmbH, Munich, DE) and Qubit® 2.0 fluorometer (Thermo Fisher Scientific, Darmstadt, Germany) according to the manufactory protocols.
The 16S rRNA gene for bacteria was amplified with the primer combination S-D-Bact-0341-b-S-17 and S-D-Bact-0785-a-A-21 (Herlemann et al., 2011). The 16S rRNA gene for archaea was amplified with the primer combination S-D-Arch-0349-a-S-17 and S-D-Arch-0786-a-A-20 (Takai and Horikoshi, 2000). The primers were labeled with unique combinations of bar codes. The PCR mix contained 1× PCR buffer (Tris ⋅ Cl, KCl, (NH4)2SO4, 15 mM MgCl2; pH 8.7) (Qiagen, Hilden, Germany), 0.5 µM of each primer (Biomers, Ulm, Germany), 0.2 mM of each deoxynucleoside (Thermo Fisher Scientific, Darmstadt, Germany), and 0.025 U µL−1 hot start polymerase (Qiagen, Hilden, Germany). The thermocycler conditions were 95 ∘C for 5 min (denaturation), followed by 40 cycles of 95 ∘C for 1 min (denaturation), 56 ∘C for 45 s (annealing), and 72 ∘C for 1 min and 30 s (elongation), concluded with a final elongation step at 72 ∘C for 10 min. PCR products were purified with a Hi Yield® Gel/PCR DNA fragment extraction kit (Süd-Laborbedarf, Gauting, Germany) according to the manufactory protocol. PCR products of three individual runs per sample were combined. PCR products of different samples were pooled in equimolar concentrations and compressed to a final volume of 10 µL with a concentration of 200 ng µL−1 in a vacuum centrifuge concentrator plus (Eppendorf, Hamburg, Germany). Individual samples were sequenced in duplicates.
The sequencing was performed on an Illumina MiSeq sequencer by the company GATC. The library was prepared with the MiSeq Reagent Kit V3 for 2×300 bp paired-end reads according to the manufactory protocols. For better performance due to different sequencing length we used 15 % PhiX control v3 library.
The quality of the sequences was checked using the fastqc tool (FastQC A Quality Control tool for High Throughput Sequence Data; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, last access: 29 June 2018; by Andrews, 2010). Raw sequence reads were demultiplexed, and bar codes were removed with the CutAdapt tool (Martin, 2011). The subsequent steps included merging of reads using overlapping sequence regions (PEAR; Zhang et al., 2014), standardizing the nucleotide sequence orientation, and trimming and filtering of low-quality sequences (Trimmomatic) (Bolger et al., 2014). After quality filtering, chimera were removed by the ChimeraSlayer tool of the QIIME pipeline. Subsequently, sequences were clustered into operational taxonomic units (OTUs) at a nucleotide cutoff level of 97 % similarity and singletons were automatically deleted. To reduce noise in the dataset, sequences with relative abundances below 0.1 % per sample were also removed. All archaeal libraries contained at least > 18 500 sequences, while bacterial libraries contained at least > 12 500 sequences. OTUs were taxonomically assigned employing the GreenGenes database 13.05 (McDonald et al., 2012) using the QIIME pipeline (Caporaso et al., 2010).
Representative sequences of OTUs were checked for correct taxonomical classification by phylogenetic tree calculations in the environment. Relative abundance of sequences related to known methanogens, anaerobic methanotrophs (ANME), and sulfate reducers were used to project microbial depth profiles. Sequences have been deposited at NCBI under the BioProject PRJNA356778 with the sequence read archive accession numbers SRR5118134-SRR5118155 for bacterial and SRR5119428-SRR5119449 for archaeal sequences, respectively.
3.1 Pore water geochemical patterns and pore water isotope composition
Substantial amounts of dissolved salts with EC maxima of up to 11.5 mS cm−1 occurred at peat depths below 30 cm b.s.f. (centimeters below surface; Fig. 2a, Table A1) and corresponded with brackish pore water proportions of up to 60 % (based on Baltic Sea salinity reported by Feistel et al., 2010). Only at spot 1, with the greatest distance to the coastline, did lower EC values (max. 3.4 mS cm−1) indicate minor brackish pore water proportions (5 %–6 %). At the other three spots, EC values were similar, i.e., exhibited no lateral salinity graduation along the remaining Baltic Sea–freshwater transect.
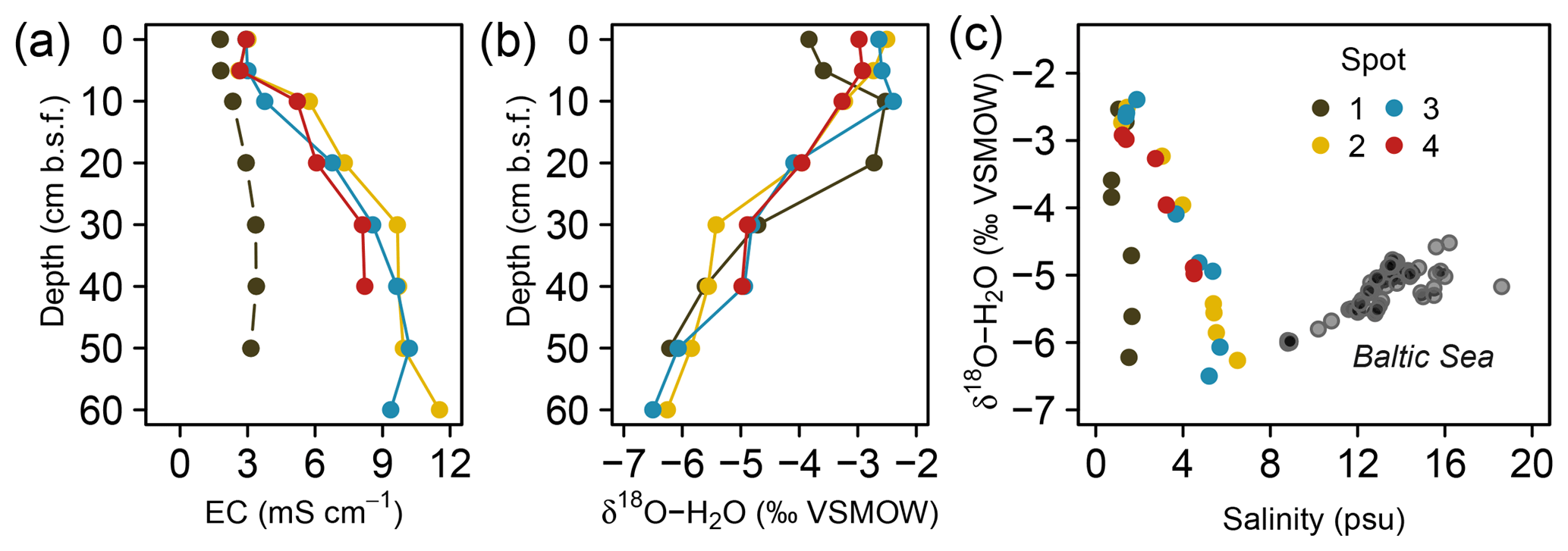
Figure 2Depth distribution of electrical conductivity (EC, a) and pore water O isotope composition (b). Panel (c) depicts a scatter plot of pore water O isotope composition and salinity. Grey transparent dots in (c) represent a common positive δ18O–H2O vs. salinity relationship derived from a sampling campaign of Baltic Sea surface water (Westphal, unpublished).
Vertical trends in pore water stable O isotope composition were similar for all spots and complementary to the salinity and EC patterns with an upwards increase from 60 to 10 cm b.s.f. (Fig. 2b). The resulting salinity–δ18O relationship was negative (except for the low salinity gradient at freshwater spot 1) and thus inverse to the common salinity–δ18O trend characteristic for Baltic coastal waters (Fig. 2c). This suggests that distribution patterns of salinity have formed independently from evaporative fractionating effects observed in the top pore water layers.
The pore water geochemistry in the peatland was increasingly diversified with depth: while the top 10 cm b.s.f. was comparatively homogenous across all spots, specific patterns evolving from diagenetic differences emerged primarily in deeper pore waters. Principal component analysis (Fig. 3) revealed the pore water geochemical composition below 10 cm b.s.f. to be constrained by two major components that evolved in opposed lateral directions and, in concert, explained 90 % of the variation in pore water composition. A distinct gradient associated with a depth increase in EC and the associated conservative ions (Cl−, Na+, Br−) suggests a persistent brackish impact at spots 2, 3, and 4 (first principal component, explained 55 % of the total variation). Only at spot 1, farthest away from the coastline, was the EC increase with depth minute. This EC gradient was further negatively correlated with pH, indicating a general decrease in pH with depth and the highest pH values around 7.0 at spot 1. A second distinct lateral gradient was delineated by the concentrations of dissolved Fe, Mn, DIC, and Ca, which occurred in higher abundances at spots 1 and 2 closest to the upstream terrestrial catchment boundary (second principal component, explained 35 % of the total variation). Such a lateral shift in pore water geochemistry is probably related to the supply of mineral solutes from terrestrial inflow. In this regard, the pore water composition of spot 2 united the elevated supply in mineral compounds from terrestrial inflow with persisting remnants of former brackish impact.
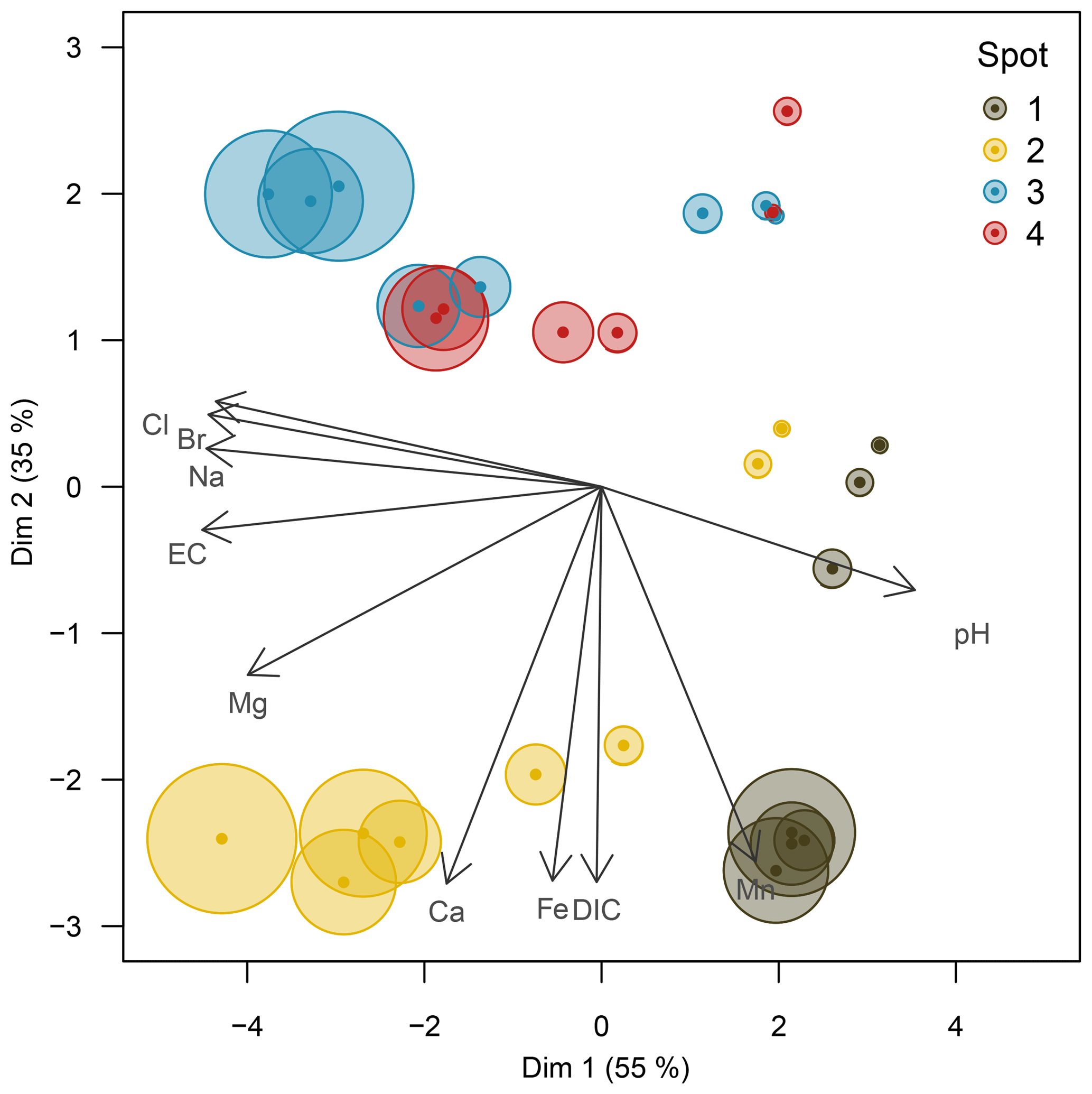
Figure 3Principal component biplot of pore water geochemical patterns within the peatland. Different colors indicate different sampling locations within the brackish–freshwater continuum with spot 1 closest to the freshwater catchment and spot 4 closest to the Baltic Sea. The size of the data points scales with sampling depth (smallest points indicate surface patterns; largest points indicate pore water composition at 60 cm in depth).
3.2 Sulfur speciation, S isotope patterns, and sulfate reducing communities
We found distinct differences in the S biogeochemical patterns across spots indicating different sulfate supply and transformation processes along the terrestrial–brackish continuum. In the following, we structured the results spot-wise according to the specific S regime and address first spot 1 (low solid sulfur and low sulfate), then spots 3 and 4 (high solid sulfur and low sulfate), and finally spot 2 (high solid sulfur and partially high sulfate concentrations).
3.2.1 Spot 1
Spot 1 characterized by low salinities and mineral inflow from the near freshwater catchment exhibited the lowest sulfate concentrations of ≤0.3 mM. H2S concentrations hardly exceeded the detection limit (∼1 µM, Fig. 4). Sulfate made up only a small proportion of the TSdiss pool, thereby indicating a higher abundance of a non-specified dissolved S fraction, probably composed of dissolved organic S, polysulfides, and S intermediates.
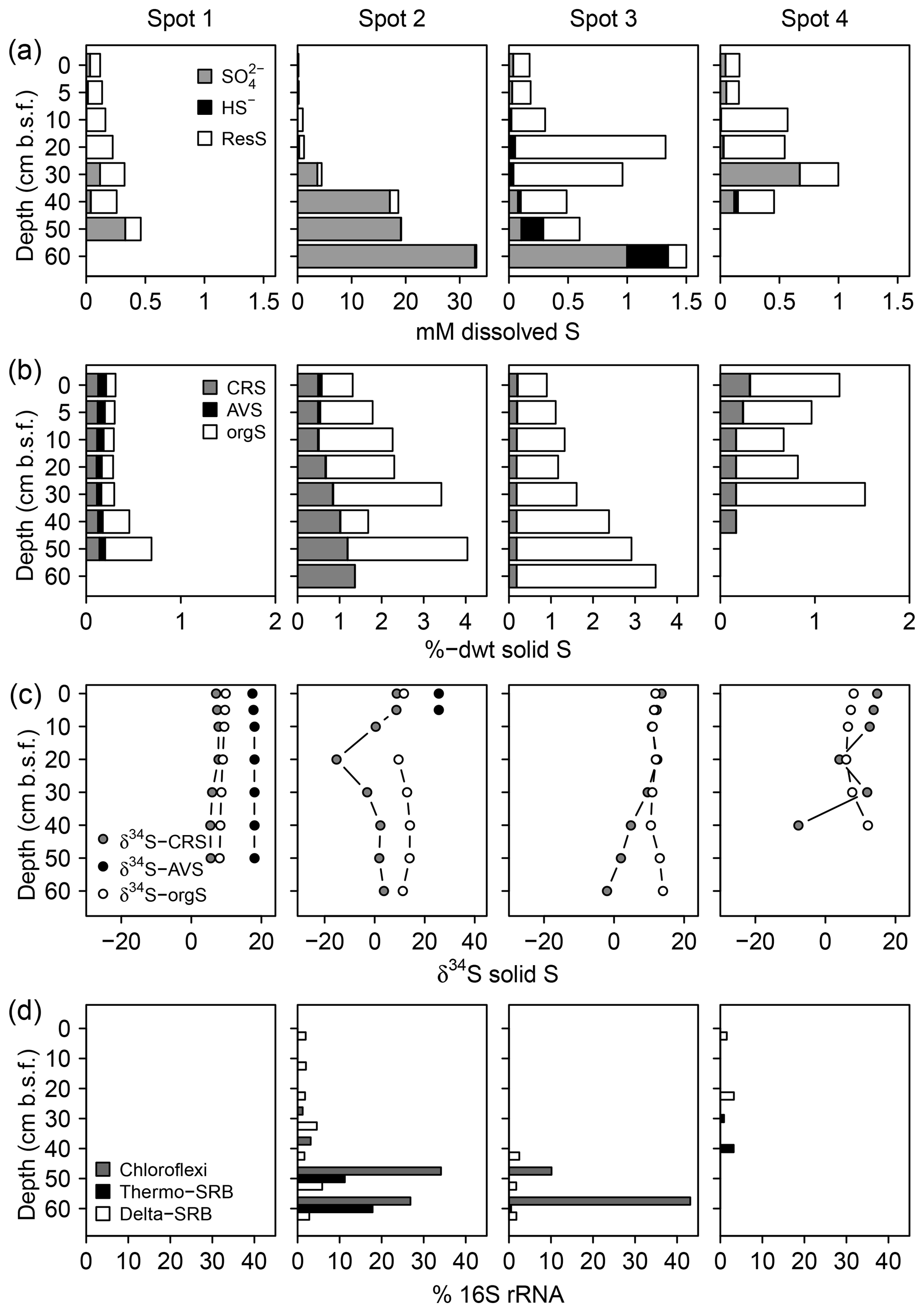
Figure 4Speciation of dissolved (a) and solid (b) S compounds, S isotope composition of solid S compounds (c), and average relative abundances of sulfate-reducing bacteria (SRB, d). δ34S and δ18O ratios of are displayed in Fig. 6a. The residual dissolved S (ResS in a) refers to a non-specified S fraction resulting from the difference between total dissolved S, H2S, and . ResS is most likely composed of dissolved organic S, polysulfides, and S intermediates. Solid S fractions (b) include iron monosulfide operationally defined as acid volatile sulfur (AVS), pyrite extracted as chromium-reducible sulfur (CRS), and a residual fraction suggested to consist primarily of organic S (orgS). δ34S at AVS could only be measured at spot 1 and the top of spot 2. SRB were extracted from two replicates of the 16S rRNA bacterial community sequencing and are assigned to the Deltaproteobacteria (Delta-SRB) and the Nitrospirae phyla (genus Thermodesulfovibrionaceae – Thermo-SRB). Chloroflexi Dehalococcoides (Chloroflexi) have not been assigned to SRB in the classical sense; however, they could be potentially involved in S metabolism (Wasmund et al., 2016). Note the different x axis scales.
In addition, the abundance of solid S was lowest at spot 1 (≤0.7 %dry weight (dwt) TSsolid). Among solid S compounds, organic-bond S constituted the dominant solid S fraction (0.1 to 0.5 %dwt) with relatively stable δ34S ratios (+8.1 ‰ and +9.8 ‰). Pyrite contents (measured as CRS) were low despite abundant pore water Fe and available solid iron (Fig. 5). Only at spot 1 did we find a low though consistent abundance of iron monosulfides (0.1 %dwt, measured as AVS). Biogeochemical turnover processes here might operate under sulfate-limited conditions resulting in lower sedimentary S contents and accumulation of iron monosulfides.
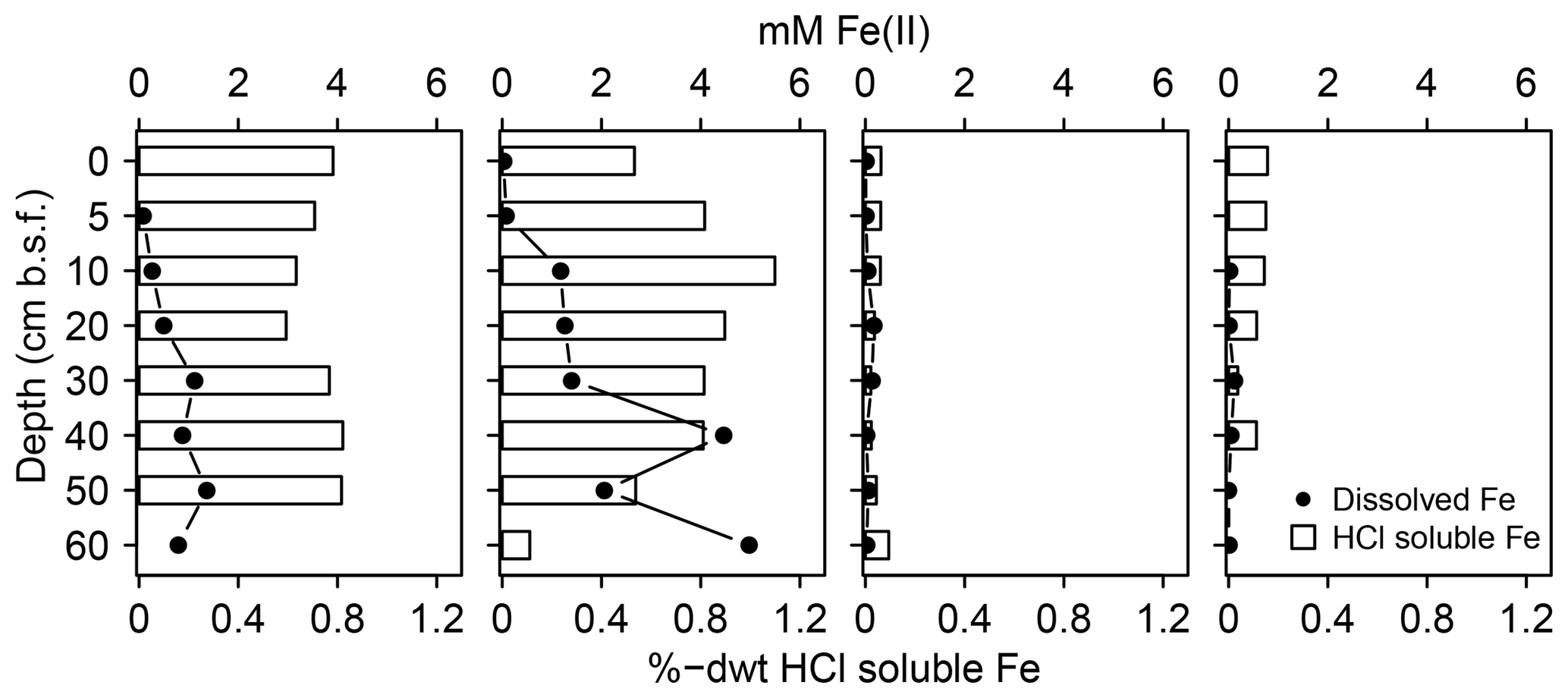
Figure 5Mobile Fe species. Available solid iron was extracted as HCl soluble iron from the sediment matrix and is composed of iron mono-sulfide and non-sulfidized ferric Fe.
In correspondence with the low sulfate contents, no sulfate-reducing bacteria occurred at spot 1.
3.2.2 Spots 3 and 4
Despite the persisting brackish impact found in the deeper pore waters of spots 3 and 4 closest to the Baltic Sea, we found hardly any pore water sulfate in the top 20 cm b.s.f. (≤0.1 mM) and only moderate levels down to 30 cm b.s.f. (0.1–1 mM). H2S abundance was essentially restricted to the depth at spot 3 (up to 347 µM).
Low pore water sulfate concentrations prevented δ34S measurements at the majority of the data points. However, the single δ34S value of +86.4 ‰ measured at 60 cm b.s.f. of spot 3 (Fig. 6a) indicated a remarkable 34S enrichment in relation to Baltic Sea water (+21 ‰; Böttcher et al., 2007). Sulfur isotope fractionation to this extent is likely to result from a superposition of enzymatic kinetic fractionation associated with a reservoir effect and constitutes striking isotopic evidence for the exhaustion of the brackish sulfate pool by intense DSR (Hartmann and Nielsen, 2012). Despite missing isotope measurements, it is likely that the low sulfate concentrations at the remaining depth sections of spot 3 and along the depth profile of spot 4 result from the same intense sulfate reduction processes.
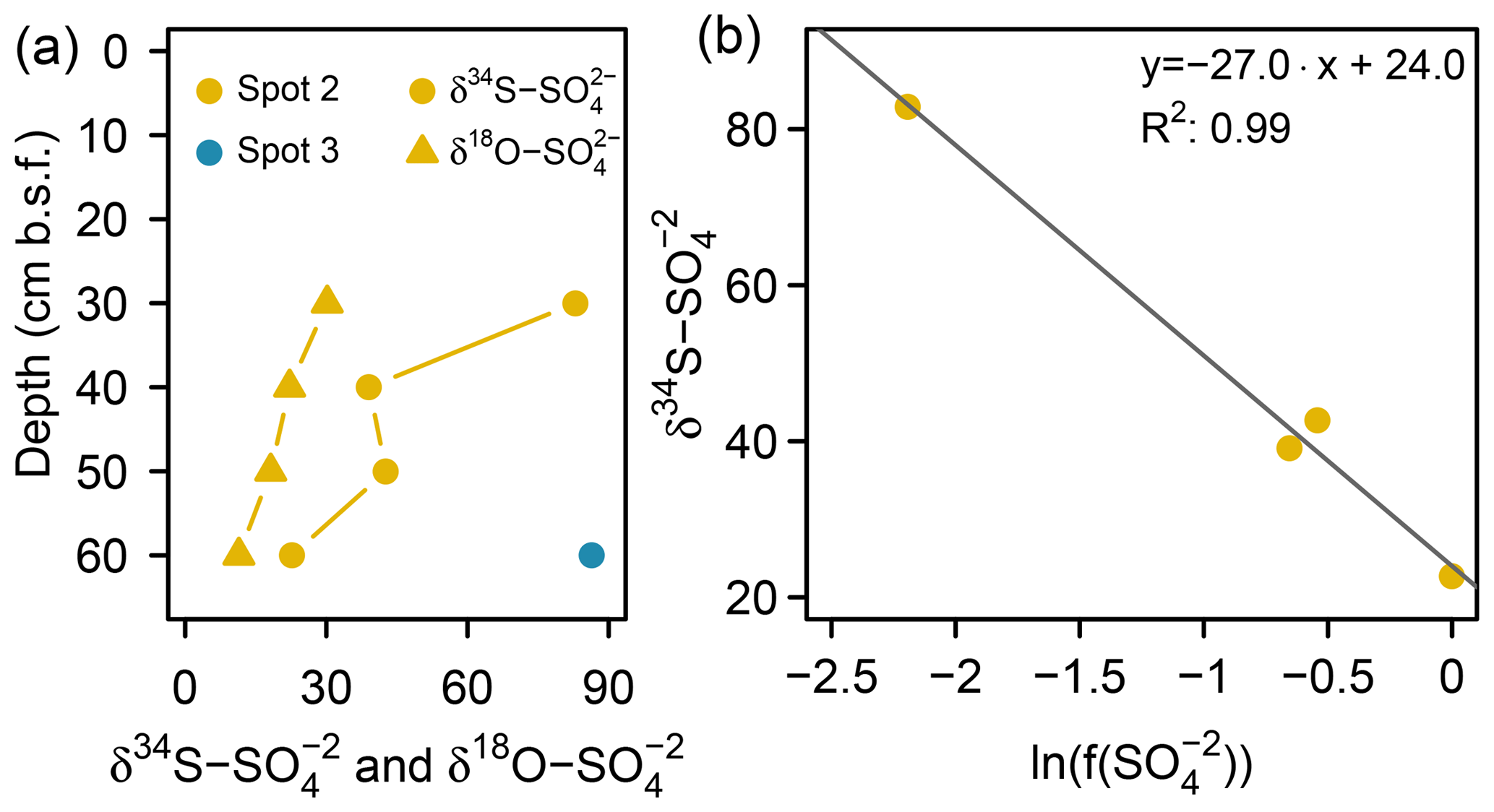
Figure 6(a) S and O isotope composition of sulfate. Sufficient for δ34S and δ18O ratio analysis was only available at the bottom of spot 2 and spot 3 (here only δ34S). (b) Rayleigh plot for measured depletion at spot 2.
We measured high amounts of TSsolid (up to 3.5 %dwt) at the depth of spot 3. In both, spots 3 and 4, organic-bond S constituted the dominant solid S fraction (0.5 to 3.3 %dwt) but was completely missing at the depth of spot 4. Pyrite was less abundant (0.2–0.3 %dwt) and exhibited a wide range of δ34S ratios (−15 ‰ to +11 ‰). As pyrite δ34S ratios essentially reflect the isotopic signature of the sulfide pool derived from DSR (Butler et al., 2004; Price and Shieh, 1979), the found variation in pyrite δ34S ratios reflected different stages of a reservoir effect that varies in response to the openness of the system (i.e., connectivity to the sea).
In correspondence with the exhaustion of the brackish sulfate pool, the relative abundance of SRB was generally small (< 5 %) and most likely substrate-limited. SRB were from the Deltaproteobacteria class and the Thermodesulfovibrionaceae genus of the Nitrospirae phylum. With 40 % relative abundance, Chloroflexi of the class Dehalococcoidetes represented the dominating bacterial group at the 1 mM concentration depth of spot 3.
3.2.3 Spot 2
At spot 2 – the interface between brackish impact and mineral inflow from the freshwater catchment – we found a sharp rise in concentration from ≤0.3 mM at the top 20 cm up to 32.8 mM at 60 cm b.s.f. The latter exceeded the quantities expected from marine supply (Kwiecinski, 1965; Feistel et al., 2010) by a factor of 8. The pronounced concentration gradient at spot 2 was associated with a remarkable variation in the stable isotope composition showing a downcore decrease in δ34S– from +82.9 ‰ to +22.7 ‰ and a decrease in δ18O– from +30 ‰ to +11 ‰ (Fig. 6a). δ34S values > +80 ‰ at 30 cm b.s.f. of spot 2 suggest the brackish sulfate pool in the top pore waters to be microbially exhausted under the same reservoir effect as in spots 3 and 4. The δ18O and δ34S ratios of excess in 60 cm b.s.f. (δ34S: +22.7 ‰; δ18O: +11.4 ‰) corresponded well with modern-day seawater (δ34S: +21 ‰; δ18O: +9 ‰; Böttcher et al., 2007). Altogether, the sharp sulfate concentration and isotope gradients at spot 2 could demonstrate the entire spectrum of sulfate speciation from the persistence of a marine sulfate reservoir at 60 cm b.s.f. towards progressing sulfate depletion in the upper peat layers.
To test this hypothesis, we applied a closed-system (Rayleigh-type) model (Eq. 1, Mariotti et al., 1981) to the data from spot 2 and gained an estimate for the δ34S ratios of the initial reservoir () and the kinetic isotope enrichment factor ε:
Here represents the S isotope values measured in specific depths of spot 2, and constitutes the fraction of remaining pore water in relation to the initial sulfate reservoir (32.8 mM , measured in 60 cm b.s.f. at spot 2). The fit through four data points (R2: 0.99; p > 0.05) revealed the δ34S ratios of the initial reservoir (+24 ‰) to be close to the 34S signature of the Baltic Sea (Fig. 6b). The isotopic offset is within the uncertainty of the estimate. The isotope enrichment factor ε was estimated to be −27 ‰, which is within the range reported for DSR in laboratory studies with pure cultures (Kaplan and Rittenberg, 1964; Canfield, 2001; Sim et al., 2011) and in the field (Habicht and Canfield, 1997; Böttcher et al., 1998).
The pronounced sulfate distribution patterns at spot 2 went along with the highest amounts of pyrite (0.5–1.4 %dwt). Pyrite contents increased with depth and partially exceeded the amounts of organic-bond S. The patterns in pyrite δ34S ratios did not correspond with the vertical trend in sulfate availability. Instead, δ34S values were lowest in 20 cm b.s.f. (−15 ‰) and stabilized around +2 ‰ below.
Interestingly, at peak sulfate supply of spot 2, the relative abundance of Deltaproteobacteria did not exceed 5 %. Instead, the SRB community at depth was dominated by the Thermodesulfovibrionaceae genus that contributed up to 21 % of all bacterial 16S rRNA sequences. Likewise with spot 3, Chloroflexi of the class Dehalococcoidetes also represented the dominating bacterial group at the depth of spot 2.
3.3 Dissolved methane concentrations, isotopic signature, and methanogenic communities
Measured pore water CH4 concentrations were up to 643 µM with equivocal vertical patterns across spots (Fig. 7a), reflecting the methane-specific spatial variability that evolves from small-scale heterogeneity in production and consumption processes and from ebullitive release events (Chanton et al., 1989; Whalen, 2005). Here, we use the isotope composition of CH4 (Fig. 7b) and DIC (Fig. 7c) to provide a clearer (and probably more robust) indication for patterns of methanogenesis and methanotrophy. Methanogenesis is a highly fractionating process: in comparison to the starting organic material (δ13C ‰ in this study), the produced CH4 is distinctively 13C-depleted, whilst at the same time, CO2 becomes considerably enriched in 13C (Whiticar et al., 1986). In this respect, high δ13C-DIC ratios up to +4.2 ‰ suggest intense methanogenic (i.e., 13C-DIC fractionating) processes in 20–40 cm b.s.f., whereas DIC on top was comparatively depleted in 13C as is characteristic for methane oxidation in the aerated surface layers. δ13C-DIC ratios below 40 cm b.s.f. converged towards the isotopic signature of bulk organic C (−26 ‰).
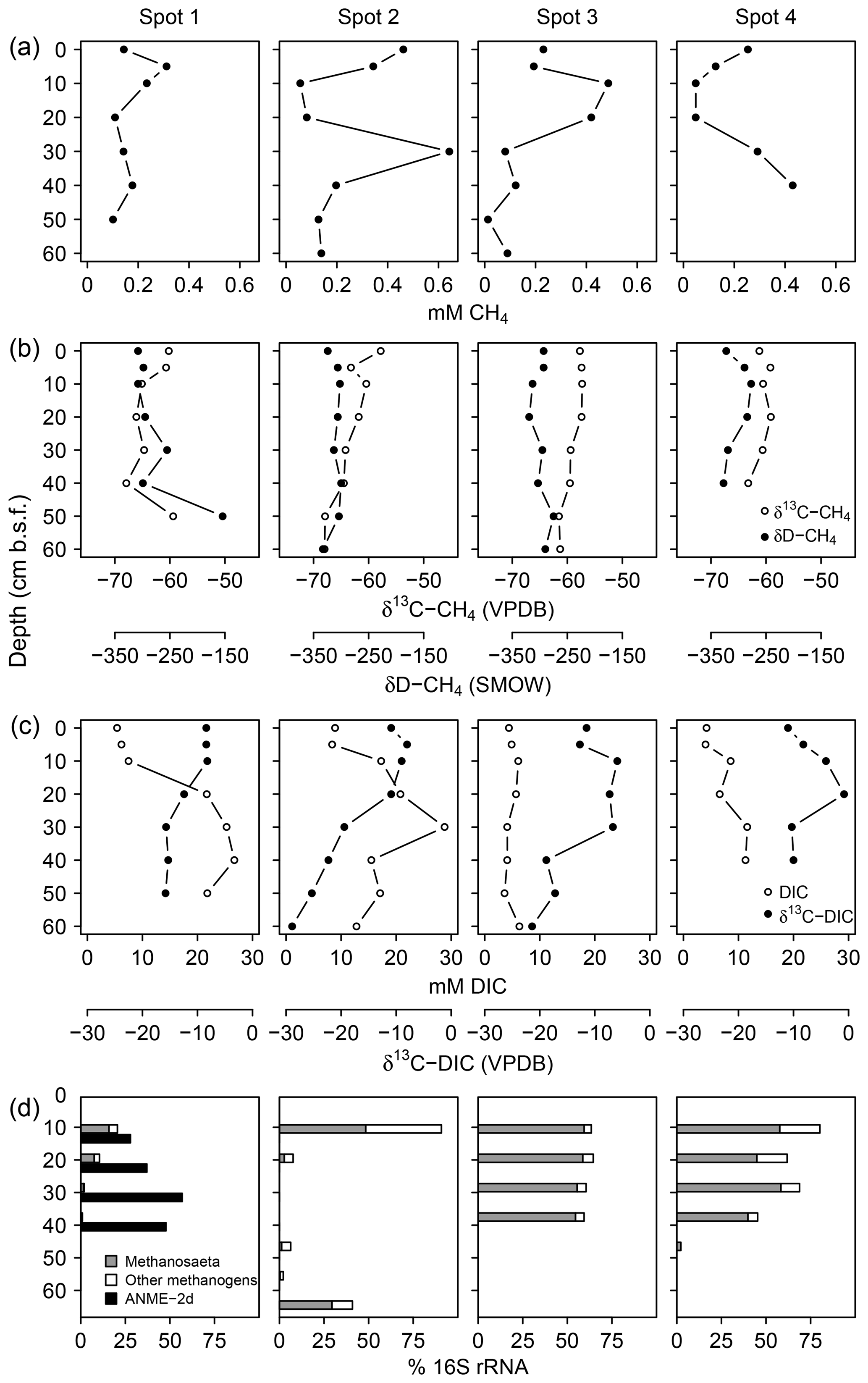
Figure 7Concentration patterns and isotope ratios for CH4 (a, b) and DIC (c), as well as average relative abundances of methanogens and methanotrophs (d).
At spot 2, we found the most pronounced downward drop in δ13C-DIC ratios with a minimum of −23.9 ‰ in 60 cm b.s.f. This pattern coincided with a consistent downward decrease in δ13C-CH4 ratios from −57 ‰ to −68 ‰ and suggests that methanogenesis operates under higher 13C fractionation associated with thermodynamically less favorable conditions at the bottom of spot 2. δD ratios of methane did not exhibit a concurrent increase but varied unrelated to δ13C-CH4 ratios in a range between −333 ‰ and −275 ‰. Based on the C and D isotopic ratio threshold raised by Whiticar (1986), acetate fermentation revealed to be the dominant methane production pathway at our study site (Fig. 8). A concurrent rise in both δD-CH4 and δ13C-CH4 ratios at the depth of spot 1 suggests a shift towards dominating CO2 reduction and/or an increase in methanotrophy.
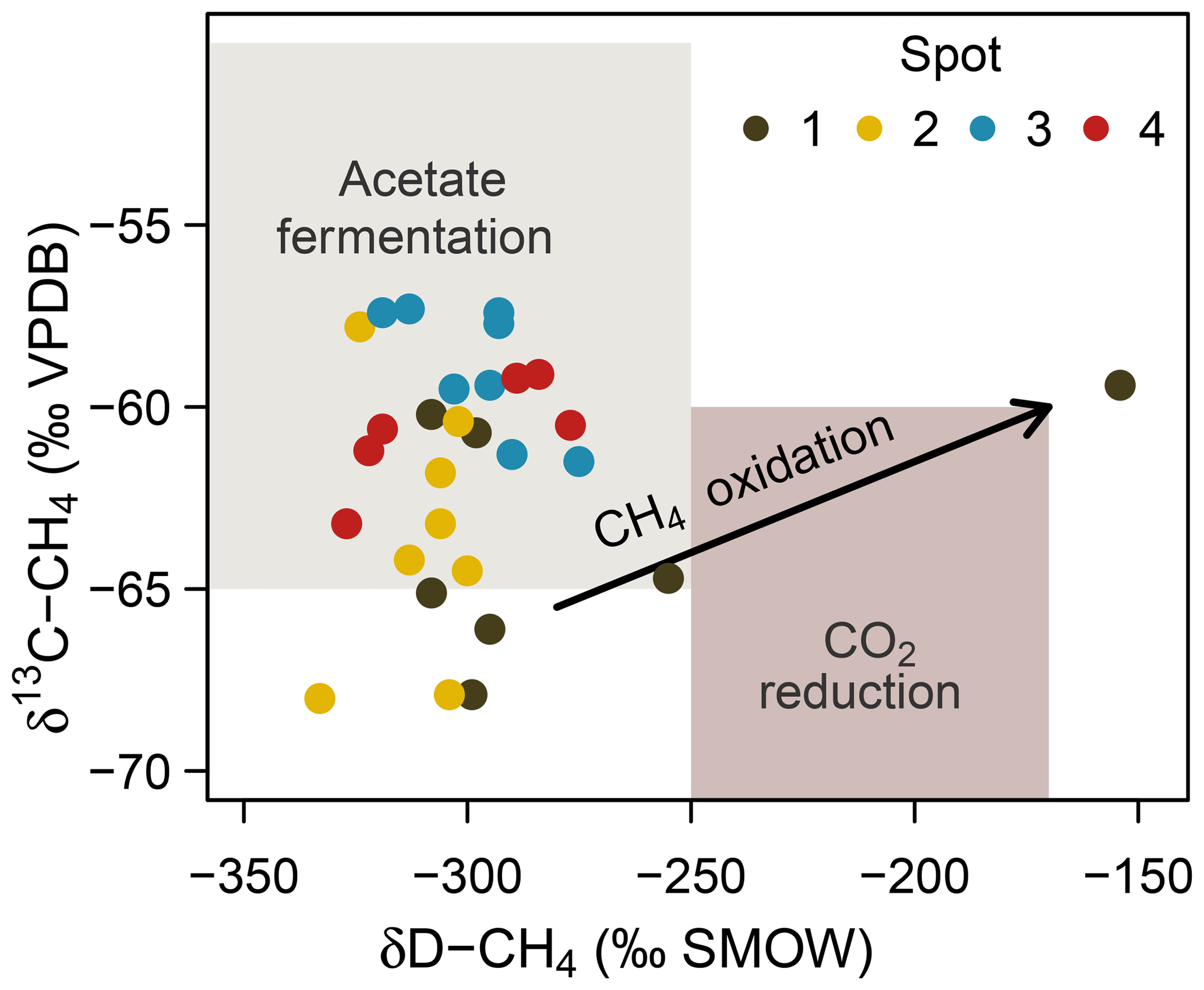
Figure 8Projection of the CH4 stable isotope composition to differentiate dominating methanogenic pathways and methanotrophy. Isotope thresholds to confine methanogenic pathways are based on Whiticar et al. (1986). The concurrent increase in δ13C-CH4 and δD-CH4 values at spot 1 suggests a downward shift towards increasing CO2 reduction or CH4 oxidation rates at depth.
Together with high δ13C-DIC ratios in the upper parts of the peat, 16S rRNA sequences related to methanogens (Fig. 7d) provided further evidence for intensive methane production. At spot 2, we found the largest divergence with 90 % methanogen-related sequences at the surface while in deeper regions (10–50 cm b.s.f.) less than 7 % of the archaeal domain could be attributed to methanogens. Surprisingly, at 60 cm b.s.f. of spot 2, methanogen percentages increased abruptly up to 41 % despite high relative abundances of SRB. Spot 1 exhibited the lowest methanogen proportions, which decreased from 21 % at the top down to 1 % in 50 cm b.s.f.
The methanogen community was mostly dominated by Methanosaeta, an obligate acetotrophic archaea genus that thrives in terrestrial organic-rich environments. The Methanosaeta proportion usually scaled with the methanogen percentage and contributed 70 %–100 % to the methanogenic community. Whilst methanogenic pathways derived from the isotopic composition of CH4 can be obscured by the fractionating effect of methanotrophy, the phylogenetic structure of the methanogenic community provided clear evidence for acetate fermentation as the prevailing methanogenic pathway in most of the peatland.
Sequences related to aerobic methanotrophs of the genus Methylosinus were only found at 30 cm b.s.f. in spot 4 representing approximately 1.5 % of all bacterial sequences (data not shown). Aerobic methanotrophs were underrepresented in our dataset.
Consistent with the concurrent depth increase in δ13C-CH4 and δD-CH4, spot 1 (Fig. 8), situated at the fringe of the freshwater catchment, exhibited high abundances of anaerobic methanotrophs of the ANME-2d clade that are so far implicated to use (Raghoebarsing et al., 2006) and/or Fe(III) (Ettwig et al., 2016) as electron acceptors.
4.1 Pore water biogeochemical patterns
Overall, the pore water geochemistry of the Hütelmoor was characterized by two different aspects: a legacy effect delineated by the lateral brackish–terrestrial continuum below 20 to 30 cm in depth and an overlying recent layer representing the uniform freshwater regime induced by rewetting.
Despite a continuous groundwater inflow from the forested catchment (Miegel et al., 2016), relics of former brackish and mineral terrestrial inflow are preserved in the deeper layers of the peat body. This is exemplified by high pore water EC values that exceeded those reported directly after the last brackish water intrusion event in 1995 (Bohne and Bohne, 2008). In fact, discharge within the peatland is channeled through rapid flow in the drainage ditches while water movement within the interstitial peat body seems to be mostly restricted to vertical exchange processes (evaporation, precipitation) with minor lateral flow (Selle et al., 2016). Therefore, we assume that drainage-induced hydrological alterations reinforced the segregation of the peat pore matrix from subsurface lateral exchange. This would allow for the preservation of residual signals in deeper pore waters and would further confine contemporary biogeochemical transformation processes to the recycling of autochthonous matter. The new top freshwater layer, established after flooding in 2010, overprints lateral differences along the brackish–fresh continuum and unifies the upper pore water geochemistry in the entire peatland.
4.2 Sulfur transformation
Along the entire brackish–terrestrial transect, virtually no sulfate was abundant in the newly developed fresh pore water layer at the top 20 cm. However, distinct differences in sulfur speciation across spots were preserved below 20 cm b.s.f. and seemed to reflect the gradual exposure to former brackish intrusion and terrestrial inflow.
Spot 1 appeared to be virtually unaffected by any brackish impact with biogeochemical turnover processes operating under sulfate-limited conditions. Low sedimentary S contents and the accumulation of iron monosulfides as representative for freshwater environments are strong points for this conclusion.
Also at spots 3 and 4, contemporary biogeochemical processes essentially operated under sulfate-limited conditions, although these areas had been exposed to flooding from the nearby Baltic Sea. High sedimentary S concentrations in conjunction with the 34S composition of the remaining sulfate suggest that the brackish sulfate reservoir has been essentially exhausted through DSR with the produced sulfide being either incorporated as diagenetically derived S in organic compounds or precipitated as 34S-enriched pyrite minerals (Brown and MacQueen, 1985; Hartmann and Nielsen, 2012). Hence, if diking of coastal wetlands prevents the replenishment of the brackish sulfate reservoir, the latter can be almost completely consumed through DSR as has been demonstrated by the Rayleigh distillation model. The rapid exhaustion of the brackish sulfate reservoir is likely to be reinforced in coastal peatlands where vast amounts of C compounds constitute an extensive electron donor supply for DSR.
Prevalent sulfate limitation at spots 1, 3, and 4 was reflected by the virtual absence of the sulfate-reducing microbial community. Interestingly, minor remnants of the brackish sulfate pool (1 mM ) at the depth of spot 3 were associated with 40 % relative abundance of Chloroflexi of the class Dehalococcoidetes. Genomes of this group in marine sediments have been shown to code for dsrAB genes (Wasmund et al., 2016). Through their ability to reduce sulfite they may be involved in S redox cycling. Indeed, further research is required to better establish their function in the S cycle.
S geochemistry at spot 2, which unites the effects of brackish water intrusion with mineral inflow of terrestrial origin, differed substantially from the other spots with remarkably high sulfate concentrations (33 mM) at depth. The mineral impact from terrestrial inflow was not only reflected by high concentrations of dissolved constituents (Fe, DIC, Mg, Ca, Mn) but also by high contents of labile iron minerals and dissolved ferrous iron. Interactions with poorly ordered ferric hydroxides can supply Fe(III) as a competitive electron acceptor next to sulfate (Postma and Jakobsen, 1996) and may, therefore, inhibit the efficient microbial reduction of the brackish sulfate reservoir. Amorphous ferric hydroxides effectively suppressed DSR in a recently rewetted Baltic coastal wetland (Virtanen et al., 2014). In our study, high contents of labile iron minerals and dissolved ferrous iron at the depth of spot 2 coincided with a high abundance of Thermodesulfovibrionaceae and a concurrently minor occurrence of Deltaproteobacteria. Recent in vitro experiments suggest Thermodesulfovibrionaceae can utilize ferric iron as an electron acceptor next to sulfate (Fortney et al., 2016). Indeed, the demonstration of Fe(III) reduction by Thermodesulfovibrionaceae under in situ conditions is currently still pending. Nevertheless, high contents of labile iron minerals, the remarkable accumulation of pore water iron, and the absence of typical iron reducers (Geobacteraceae, Peptococcaceae, Shewanellaceae, Desulfovibrionaceae, Pelobacteraceae) could suggest Thermodesulfovibrionaceae prefer Fe(III) as an electron acceptor over sulfate. Thus, the unique concentration patterns at spot 2 may be attributed to the inhibited microbial consumption of the brackish sulfate reservoir caused by the delivery of alternative electron acceptors from the nearby freshwater catchment.
Altogether, our results demonstrate the potential fate of the brackish sulfate reservoir in coastal wetlands under closed system conditions caused by diking. Microbial transformation processes have decoupled the sulfate distribution patterns from the relic brackish impact and have caused marked differences in contemporary sulfate biogeochemistry: on the one hand, DSR exhausted the brackish sulfate reservoir in wide parts of the peatlands, whereas, on the other hand, the preferential consumption of competitive electron acceptors from terrestrial origin allowed for the local accumulation of large sulfate concentrations. Indeed, these relic signals of brackish–terrestrial intermixing are constrained to the deeper pore water regions below 30 cm b.s.f. as recent rewetting measures established a homogeneous freshwater regime in the top layers of the entire peatland.
4.3 Methane production and consumption
δ13C-DIC ratios and a thriving methanogenic community indicate the establishment of distinct methane production zones in the recently formed freshwater layer across the entire peatland. In line with the prevalent freshwater characteristics of the newly formed pore water layer, the methanogen community was dominated by Methanosaeta, an obligate acetotrophic genus typical of terrestrial organic-rich environments. Indeed, thermodynamically favorable methanogenic conditions were confined to the top layers since isotopic evidence and archaeal distribution patterns indicate a downward shift towards non-fractionating metabolic processes (Barker, 1936; Lapham et al., 1999) at the bottom. This vertical transition was most pronounced at spot 2, probably indicating a potential suppression of methanogenesis by high concentrations of sulfate and labile ferric iron compounds at depth.
Surprisingly, we observed mutual coexistence of SRB (22 % of all bacterial sequences) and methanogens (> 40 % of all archaeal sequences) at high concentrations (32.8 mM) in 60 cm b.s.f. at spot 2. Simultaneous methanogenesis and DSR have been reported under the abundance of methanol, trimethylamine, or methionine as methanogenic precursors (Oremland and Polcin, 1982). However, the concurrent high abundance of Methanosaeta (30 %) at the depth of spot 2 suggests competitive consumption of acetate by both SRB and methanogens. Although Liebner et al. (2015) emphasized the relevance of community structure with regard to prevailing methanogenic pathways, total abundance data could potentially yield more insight into this issue.
Sequences related to aerobic methanotrophs of the genus Methylosinus were only found at 30 cm b.s.f. in spot 4, representing approximately 1.5 % of all bacterial sequences (data not shown). The phenomenon of a lagged reestablishment of methanotrophs in comparison to methanogens after rewetting in this particular peatland is addressed in another publication (Wen et al., 2018).
Despite the overlap of methane production zones anticipated from δ13C-DIC ratios with sulfate reduction zones, we could not find evidence for the syntrophic consortium of anaerobic methanotrophs (ANME) and sulfate reducers that is commonly associated with the anaerobic oxidation of methane coupled to sulfate reduction (AOM-SR) in marine environments (Boetius et al., 2000). However, we cannot exclude that AOM-SR is driven by archaea that are so far not known for this function. One potential candidate phylum is the Bathyarchaota that have been shown to encode an untypical version of the functional gene for methane production and consumption (methyl coenyzme M reductase subunit A, mcrA) (Evans et al., 2015). These archaea dominated spot 2 with 48 %–97 % relative sequence abundance of the archaeal community between 10 and 60 cm (data not shown).
While we cannot supply microbial evidence for AOM-SR, high abundances of anaerobic methanotrophs of the ANME-2d clade at spot 1 suggest anaerobic methane oxidation coupled to electron acceptors of terrestrial origin. Methanotrophs of the ANME-2d clade are so far known to utilize (Raghoebarsing et al., 2006) and ferric iron (Ettwig et al., 2016) as electron acceptors, both of which were abundant at the respective spot. This observation is further supported by the trend in δ13C-CH4 and δD-CH4 that potentially indicates a downward increase in methanotrophy at spot 1. The biogeochemical characteristics at this very location result most likely from formerly drier conditions due to slightly higher elevation in combination with prevalent inflow from the nearby forest catchment.
Our results demonstrate how rewetting of a coastal peatland established a distinct freshwater regime in the upper pore water layers, which, in conjunction with prevalent anaerobic conditions and a vast stock of labile C compounds, offers favorable conditions for intense methane production and explains the high methane emissions reported in Hahn et al. (2015) and Koebsch et al. (2015). As intense methane production was confined to the upper pore water layers in the entire peatland, it did not interfere with high sulfate concentrations locally preserved as the legacy of former brackish impact in the bottom. Instead, isotopic and microbial evidence suggested mineral compounds of terrestrial origin to constitute an electron acceptor for anaerobic methane oxidation, which is an often neglected – though it is an important process in freshwater environments (Segarra et al., 2015). Our results indicate that this process can also occur in disturbed coastal peatlands. Indeed, the quantitative effects of anaerobic methane consumption on methane emissions in coastal and/or rewetted peatlands need to be addressed in future studies.
In this study, we investigated the biogeochemical and hydrological mechanisms that turn disturbed and remediated coastal peatlands into strong methane sources. Our study demonstrates how human intervention overrides the sulfate-related processes that suppress methane production and thereby suspends the natural mechanisms that mitigate greenhouse gas emissions from coastal environments. Hence, the climate effect of disturbed and remediated coastal wetlands cannot simply be derived by analogy with their natural counterparts. Instead, human alterations form new transient systems where relic brackish signals intermingle with recent freshwater impacts. The evolving biogeochemical patterns overprint naturally established gradients formed, for instance, by the distance to the coastline. In particular, the decoupling of sulfate abundance from salinity is of high practical relevance for greenhouse gas inventories that establish methane emission factors based on the empirical relation to salinity as an easily accessible proxy for sulfate concentrations.
Coastal environments are subject to particular pressure by high population density while at the same time their potential as coastal buffer zones is moving more and more into the focus of policy makers and land managers. From a greenhouse gas perspective, the exposure of diked wetlands to natural coastal dynamics would literally open the floodgates for a replenishment of the marine sulfate pool and constitute an efficient measure to reduce methane emissions. However, in practice, this option has to be weighed against concurrent land use aspects.
Geochemical data are represented within this paper in the appendix (Table A1). Sequences have been deposited at NCBI under the BioProject PRJNA356778 with the sequence read archive accession numbers SRR5118134-SRR5118155 for bacterial and SRR5119428-SRR5119449 for archaeal sequences.
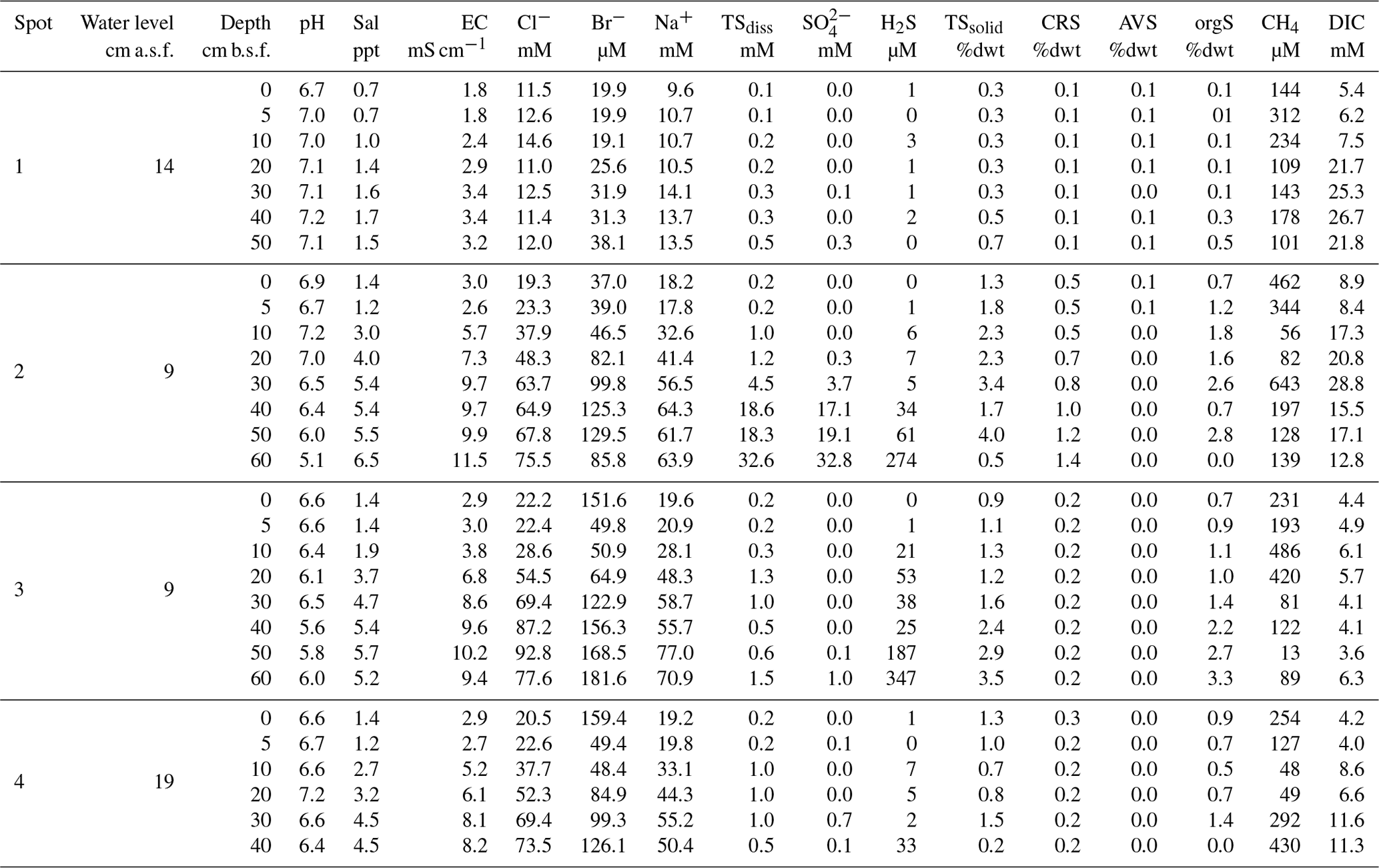
Table A1Site parameters, pore water, and soil characteristics. Water level and soil depth are given in centimeters above and centimeters below surface (cm a.s.f. and cm b.s.f., respectively).
FK and MEB have formulated the research question and planned the study design. FK acquired funding. FK, GJ, MK, MW, and SK collected the samples. MEB, SL, AS, MG, TS, and SK provided resources and lab instrumentation for sample analysis. FK, AS, IS, MK, GJ, SK, and JW conducted the geochemical analyses. MW, SL, and VU conducted the microbial sequencing analysis. BL validated the results. FK visualized the data and prepared the original draft with contributions from all coauthors.
The authors declare that they have no conflict of interest.
This work was supported by the DFG Research Training Group BALTIC TRANSCOAST (grant DFG GRK
2000) and the Helmholtz Terrestrial Environmental Observatories (TERENO) Network. This is BALTIC TRANSCOAST publication number
GRK2000/0023. Franziska Koebsch was supported by the Helmholtz Association of German
Research Centers through the Helmholtz Postdoc Program (grant PD-129) and
the Helmholtz Climate Initiative REKLIM (Regional Climate Change). Franziska Koebsch was further
supported by the European Social Fund (ESF) and the Ministry of Education,
Science and Culture of Mecklenburg-West Pomerania within the scope of the
project WETSCAPES (ESF/14-BM-A55-0030/16). Torsten Sachs and Susanne Liebner were each supported by
a Helmholtz Young Investigators Group (grants VH-NG-821 and VH-NG-919).
Biogeochemical and stable isotope work was supported by the Leibniz
Institute for Baltic Sea Research (IOW). We wish to express our gratitude to
Lisa Kretzschmann, Anke Saborowski, and Simon Strunk for their commitment to field
work under tough conditions. Bennet Juhls and Simon Strunk have helped with map
creation. The study would not have been possible without the laboratory and
bioinformatics support by Andrea Gottsche, Anke Saborowski, Lisa Kretzschmann, Anne Köhler, Birgit Plessen, Vera Winde, Fabian Horn, Xi Wen, and
Heiko Baschek.
We acknowledge financial support by Deutsche Forschungsgemeinschaft and Universität Rostock within the funding programme Open Access Publishing.
This paper was edited by Jianming Xu and reviewed by two anonymous referees.
Andrews, S.: FastQC: a quality control tool for high throughput sequence data, available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (last access: 29 June 2018), 2010.
Barker, H. A.: On the biochemistry of the methane fermentation, Arch. Mikrobiol., 7, 404–419, 1936.
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., Amann, R., Jørgensen, B. B., Witte, U., and Pfannkuche, O.: A marine microbial consortium apparently mediating anaerobic oxidation of methane, Nature, 407, 623–626, 2000.
Bohne, B. and Bohne, K.: Monitoring zum Wasserhaushalt einer auf litoralem Versumpfungsmoor gewachsenen Regenmoorkalotte– Beispiel Naturschutzgebiet “Hütelmoor” bei Rostock, Aspekte der Geoökologie, Weißensee Verlag, Berlin, Germany, 2008.
Bolger, A. M., Lohse, M., and Usadel, B.: Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–2120, 2014.
Böttcher, M., Brumsack, H.-J., and Dürselen, C.-D.: The isotopic composition of modern seawater sulfate: I. Coastal waters with special regard to the North Sea, J. Marine Syst., 67, 73–82, 2007.
Böttcher, M. E. and Lepland, A.: Biogeochemistry of sulfur in a sediment core from the west-central Baltic Sea: evidence from stable isotopes and pyrite textures, J. Marine Syst., 25, 299–312, 2000.
Böttcher, M. E., Brumsack, H.-J., De Lange, G. J., and Robertson, A.: Sulfate reduction and related stable isotope (34S, 18O) variations in interstitial waters from the Eastern Mediterranean, 365–376, 1998.
Brand, W. A. and Coplen, T. B.: Stable isotope deltas: tiny, yet robust signatures in nature, Isot. Environ. Healt. S., 48, 393–409, 2012.
Brown, K. and MacQueen, J.: Sulphate uptake from surface water by peat, Soil Biol. Biochem., 17, 411–420, 1985.
Butler, I. B., Böttcher, M. E., Rickard, D., and Oldroyd, A.: Sulfur isotope partitioning during experimental formation of pyrite via the polysulfide and hydrogen sulfide pathways: implications for the interpretation of sedimentary and hydrothermal pyrite isotope records, Earth Planet. Sc. Lett., 228, 495–509, 2004.
Canfield, D. E.: Reactive iron in marine sediments, Geochim. Cosmochim. Ac., 53, 619–632, 1989.
Canfield, D. E.: Isotope fractionation by natural populations of sulfate-reducing bacteria, Geochim. Cosmochim. Ac., 65, 1117–1124, 2001.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Pena, A. G., Goodrich, J. K., and Gordon, J. I.: QIIME allows analysis of high-throughput community sequencing data, Nat. Methods, 7, 335–336, 2010.
Chanton, J. P., Martens, C. S., and Kelley, C. A.: Gas transport from methane-saturated, tidal freshwater and wetland sediments, Limnol. Oceanogr., 34, 807–819, 1989.
Cline, J. D.: Spectrophotometric determination of hydrogen sulfide in natural waters 1, Limnol. Oceanogr., 14, 454–458, 1969.
Dahms, P.: Studie Wasserregulierung Hütelmoor, Universität Rostock, Fachbereich Landeskultur und Umweltschutz, Fachgebiet Kulturtechnik, Rostock, Germany, 1991.
Deverel, S. J. and Rojstaczer, S.: Subsidence of agricultural lands in the Sacramento-San Joaquin Delta, California: Role of aqueous and gaseous carbon fluxes, Water Resour. Res., 32, 2359–2367, 1996.
Deverel, S. J., Ingrum, T., and Leighton, D.: Present-day oxidative subsidence of organic soils and mitigation in the Sacramento-San Joaquin Delta, California, USA, Hydrogeol. J., 24, 569–586, 2016.
Erkens, G., van der Meulen, M. J., and Middelkoop, H.: Double trouble: subsidence and CO2 respiration due to 1000 years of Dutch coastal peatlands cultivation, Hydrogeol. J., 24, 551–568, 2016.
Ettwig, K. F., Zhu, B., Speth, D., Keltjens, J. T., Jetten, M. S., and Kartal, B.: Archaea catalyze iron-dependent anaerobic oxidation of methane, P. Natl. Acad. Sci. USA, 113, 12792–12796, 2016.
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., Golding, S. D., and Tyson, G. W.: Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics, Science, 350, 434–438, 2015.
Feistel, R., Weinreben, S., Wolf, H., Seitz, S., Spitzer, P., Adel, B., Nausch, G., Schneider, B., and Wright, D. G.: Density and Absolute Salinity of the Baltic Sea 2006–2009, Ocean Sci., 6, 3–24, https://doi.org/10.5194/os-6-3-2010, 2010.
Fortney, N., He, S., Converse, B., Beard, B., Johnson, C., Boyd, E. S., and Roden, E.: Microbial Fe (III) oxide reduction potential in Chocolate Pots hot spring, Yellowstone National Park, Geobiology, 14, 255–275, 2016.
Fossing, H. and Jørgensen, B. B.: Measurement of bacterial sulfate reduction in sediments: evaluation of a single-step chromium reduction method, Biogeochemistry, 8, 205–222, 1989.
Franz, D., Koebsch, F., Larmanou, E., Augustin, J., and Sachs, T.: High net CO2 and CH4 release at a eutrophic shallow lake on a formerly drained fen, Biogeosciences, 13, 3051–3070, https://doi.org/10.5194/bg-13-3051-2016, 2016.
Habicht, K. S. and Canfield, D. E.: Sulfur isotope fractionation during bacterial sulfate reduction in organic-rich sediments, Geochim. Cosmochim. Ac., 61, 5351–5361, 1997.
Hahn, J., Köhler, S., Glatzel, S., and Jurasinski, G.: Methane exchange in a coastal fen in the first year after flooding-a systems shift, PloS one, 10, e0140657, 2015.
Hartmann, M. and Nielsen, H.: δ34S values in recent sea sediments and their significance using several sediment profiles from the western Baltic Sea, Isot. Environ. Healt. S., 48, 7–32, 2012.
Hemes, K. S., Chamberlain, S. D., Eichelmann, E., Knox, S. H., and Baldocchi, D. D.: A biogeochemical compromise: The high methane cost of sequestering carbon in restored wetlands, Geophys. Res. Lett., 45, 6081–6091, 2018.
Herlemann, D. P., Labrenz, M., Jürgens, K., Bertilsson, S., Waniek, J. J., and Andersson, A. F.: Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea, ISME J., 5, 1571–1579, 2011.
Holm, G. O., Perez, B. C., McWhorter, D. E., Krauss, K. W., Johnson, D. J., Raynie, R. C., and Killebrew, C. J.: Ecosystem level methane fluxes from tidal freshwater and brackish marshes of the Mississippi River Delta: Implications for coastal wetland carbon projects, Wetlands, 36, 401–413, 2016.
Iversen, N. and Jorgensen, B. B.: Anaerobic methane oxidation rates at the sulfate-methane transition in marine sediments from Kattegat and Skagerrak (Denmark) 1, Limnol. Oceanogr., 30, 944–955, 1985.
Kaplan, I. and Rittenberg, S.: Microbiological fractionation of sulphur isotopes, Microbiology, 34, 195–212, 1964.
Karstens, S., Buczko, U., Jurasinski, G., Peticzka, R., and Glatzel, S.: Impact of adjacent land use on coastal wetland sediments, Sci. Total Environ., 550, 337–348, 2016.
King, G. M. and Wiebe, W.: Regulation of sulfate concentrations and methanogenesis in salt marsh soils, Estuar. Coast. Mar. Sci., 10, 215–223, 1980.
Koch, M., Koebsch, F., Hahn, J., and Jurasinski, G.: From meadow to shallow lake: Monitoring secondary succession in a coastal fen after rewetting by flooding based on aerial imagery and plot data, Mires Peat, 19, 1–17, 2017.
Koebsch, F., Glatzel, S., and Jurasinski, G.: Vegetation controls methane emissions in a coastal brackish fen, Wetl. Ecol. Manag., 21, 323–337, 2013.
Koebsch, F., Jurasinski, G., Koch, M., Hofmann, J., and Glatzel, S.: Controls for multi-scale temporal variation in ecosystem methane exchange during the growing season of a permanently inundated fen, Agr. Forest Meteorol., 204, 94–105, 2015.
Kornexl, B. E., Werner, R. A., and Gehre, M.: Standardization for oxygen isotope ratio measurement – still an unsolved problem, Rapid Commun. Mass Sp., 13, 1248–1251, 1999.
Kowalski, N., Dellwig, O., Beck, M., Grunwald, M., Dürselen, C.-D., Badewien, T. H., Brumsack, H.-J., van Beusekom, J. E., and Böttcher, M. E.: A comparative study of manganese dynamics in the water column and sediments of intertidal systems of the North Sea, Estuar. Coast. Shelf S., 100, 3–17, 2012.
Kwiecinski, B.: The sulfate content of Baltic water and its relation to the chlorinity, Deep-Sea Res., 12, 797–804, 1965.
Lapham, L., Proctor, L., and Chanton, J.: Using respiration rates and stable carbon isotopes to monitor the biodegradation of orimulsion by marine benthic bacteria, Environ. Sci. Technol., 33, 2035–2039, 1999.
Liebner, S., Ganzert, L., Kiss, A., Yang, S., Wagner, D., and Svenning, M. M.: Shifts in methanogenic community composition and methane fluxes along the degradation of discontinuous permafrost, Front. Microbiol., 6, 1–10, 2015.
Lovley, D. R. and Klug, M. J.: Sulfate reducers can outcompete methanogens at freshwater sulfate concentrations, Appl. Environ. Microb., 45, 187–192, 1983.
Mann, J. L., Vocke Jr., R. D., and Kelly, W. R.: Revised δ34S reference values for IAEA sulfur isotope reference materials S-2 and S-3, Rapid Commun. Mass Sp., 23, 1116–1124, 2009.
Mariotti, A., Germon, J. C., Hubert, P., Kaiser, P., Letolle, R., Tardieux, A., and Tardieux, P.: Experimental determination of nitrogen kinetic isotope fractionation: some principles; illustration for the denitrification and nitrification processes, Plant Soil, 62, 413–430, 1981.
Martens, C. S. and Berner, R. A.: Methane production in the interstitial waters of sulfate-depleted marine sediments, Science, 185, 1167–1169, 1974.
Martin, M.: Cutadapt removes adapter sequences from high-throughput sequencing reads, EMBnet. Journal, 17, 10–12, 2011.
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., Andersen, G. L., Knight, R., and Hugenholtz, P.: An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea, ISME J., 6, 610–618, 2012.
Miegel, K., Graeff, T., Selle, B., Salzmann, T., Franck, C., and Bronstert, A.: Untersuchung eines renaturierten Niedermoores an der mecklenburgischen Ostseeküste – Teil I: Systembeschreibung und hydrologische Grundcharakterisierung, Hydrol. Wasserbewirts., 60, 242–258; https://doi.org/10.5675/HyWa_2016,4_1, 2016.
Miller, R. L.: Carbon gas fluxes in re-established wetlands on organic soils differ relative to plant community and hydrology, Wetlands, 31, 1055–1066, 2011.
Oremland, R. S. and Polcin, S.: Methanogenesis and sulfate reduction: competitive and noncompetitive substrates in estuarine sediments, Appl. Environ. Microb., 44, 1270–1276, 1982.
Passier, H. F., Böttcher, M. E., and De Lange, G. J.: Sulphur enrichment in organic matter of eastern Mediterranean sapropels: a study of sulphur isotope partitioning, Aquat. Geochem., 5, 99–118, 1999.
Postma, D. and Jakobsen, R.: Redox zonation: equilibrium constraints on the Fe(III)∕SO4-reduction interface, Geochim. Cosmochim. Ac., 60, 3169–3175, 1996.
Price, F. T. and Shieh, Y.: Fractionation of sulfur isotopes during laboratory synthesis of pyrite at low temperatures, Chem. Geol., 27, 245–253, 1979.
Raghoebarsing, A. A., Pol, A., Van de Pas-Schoonen, K. T., Smolders, A. J., Ettwig, K. F., Rijpstra, W. I. C., Schouten, S., Damsté, J. S. S., den Camp, H. J. O., and Jetten, M. S.: A microbial consortium couples anaerobic methane oxidation to denitrification, Nature, 440, 918–921, 2006.
Sánchez-Arcilla, A., García-León, M., Gracia, V., Devoy, R., Stanica, A., and Gault, J.: Managing coastal environments under climate change: Pathways to adaptation, Sci. Total Environ., 572, 1336–1352, 2016.
Schönheit, P., Kristjansson, J. K., and Thauer, R. K.: Kinetic mechanism for the ability of sulfate reducers to out-compete methanogens for acetate, Arch. Microbiol., 132, 285–288, 1982.
Segarra, K., Schubotz, F., Samarkin, V., Yoshinaga, M., Hinrichs, K., and Joye, S.: High rates of anaerobic methane oxidation in freshwater wetlands reduce potential atmospheric methane emissions, Nat. Commun., 6, 7477–7488, 2015.
Segarra, K. E., Comerford, C., Slaughter, J., and Joye, S. B.: Impact of electron acceptor availability on the anaerobic oxidation of methane in coastal freshwater and brackish wetland sediments, Geochim. Cosmochim. Ac., 115, 15–30, 2013.
Segers, R. and Kengen, S.: Methane production as a function of anaerobic carbon mineralization: a process model, Soil Biol. Biochem., 30, 1107–1117, 1998.
Selle, B., Graeff, T., Salzmann, T., Oswald, S. E., Walther, M., and Miegel, K.: Untersuchung eines renaturierten Mooreinzugsgebiets an der mecklenburgischen Ostseeküste – Teil II: Salzdynamik und Wasserhaushalt, Hydrol. Wasserbewirts., 60, 259–268, https://doi.org/10.5675/HyWa_2016,4_2, 2016.
Sim, M. S., Bosak, T., and Ono, S.: Large sulfur isotope fractionation does not require disproportionation, Science, 333, 74–77, 2011.
Stookey, L. L.: Ferrozine – a new spectrophotometric reagent for iron, Anal. Chem., 42, 779–781, 1970.
Takai, K. and Horikoshi, K.: Rapid detection and quantification of members of the archaeal community by quantitative PCR using fluorogenic probes, Appl. Environ. Microb., 66, 5066–5072, 2000.
Vanselow-Algan, M., Schmidt, S. R., Greven, M., Fiencke, C., Kutzbach, L., and Pfeiffer, E.-M.: High methane emissions dominated annual greenhouse gas balances 30 years after bog rewetting, Biogeosciences, 12, 4361–4371, https://doi.org/10.5194/bg-12-4361-2015, 2015.
Virtanen, S., Simojoki, A., Hartikainen, H., and Yli-Halla, M.: Response of pore water Al, Fe and S concentrations to waterlogging in a boreal acid sulphate soil, Sci. Total Environ., 485, 130–142, 2014.
Voigtländer, U., Schmidt, J., and Scheller, W.: Pflege- und Entwicklungsplan NSG Heiligensee und Hütelmoor, 1996.
Wasmund, K., Cooper, M., Schreiber, L., Lloyd, K. G., Baker, B. J., Petersen, D. G., Jørgensen, B. B., Stepanauskas, R., Reinhardt, R., and Schramm, A.: Single-cell genome and group-specific dsrAB sequencing implicate marine members of the class Dehalococcoidia (Phylum Chloroflexi) in Sulfur Cycling, MBio, 7, 1–15, e00266-00216, 2016.
Wen, X., Unger, V., Jurasinski, G., Koebsch, F., Horn, F., Rehder, G., Sachs, T., Zak, D., Lischeid, G., Knorr, K.-H., Böttcher, M. E., Winkel, M., Bodelier, P. L. E., and Liebner, S.: Predominance of methanogens over methanotrophs in rewetted fens characterized by high methane emissions, Biogeosciences, 15, 6519–6536, https://doi.org/10.5194/bg-15-6519-2018, 2018.
Whalen, S.: Biogeochemistry of methane exchange between natural wetlands and the atmosphere, Environ. Eng. Sci., 22, 73–94, 2005.
Whiticar, M. J., Faber, E., and Schoell, M.: Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation – isotope evidence, Geochim. Cosmochim. Ac., 50, 693–709, 1986.
Wilhelm, E., Battino, R., and Wilcock, R. J.: Low-pressure solubility of gases in liquid water, Chem. Rev., 77, 219–262, 1977.
Wilson, D., Alm, J., Laine, J., Byrne, K. A., Farrell, E. P., and Tuittila, E. S.: Rewetting of cutaway peatlands: are we re-creating hot spots of methane emissions?, Restor. Ecol., 17, 796–806, 2009.
Winde, V., Böttcher, M., Escher, P., Böning, P., Beck, M., Liebezeit, G., and Schneider, B.: Tidal and spatial variations of DI13C and aquatic chemistry in a temperate tidal basin during winter time, J. Marine Syst., 129, 396–404, 2014.
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A.: PEAR: a fast and accurate Illumina Paired-End reAd merger, Bioinformatics, 30, 614–620, 2014.
Zhang, Z., Zimmermann, N. E., Stenke, A., Li, X., Hodson, E. L., Zhu, G., Huang, C., and Poulter, B.: Emerging role of wetland methane emissions in driving 21st century climate change, P. Natl. Acad. Sci. USA, 114, 9647–9652, 2017.
Zhao, Q., Bai, J., Huang, L., Gu, B., Lu, Q., and Gao, Z.: A review of methodologies and success indicators for coastal wetland restoration, Ecol. Indic., 60, 442–452, 2016.